Téma: Parathormon a poruchy jeho sekrece a účinku
Posted By vodouch On 7.9.2012 @ 7:20 In 5.2. Poruchy iontové rovnováhy | Comments Disabled
Pracoviště: Ústav patologické fyziologie LF UP Olomouc
Úvod
Syntéza a sekrece PTH
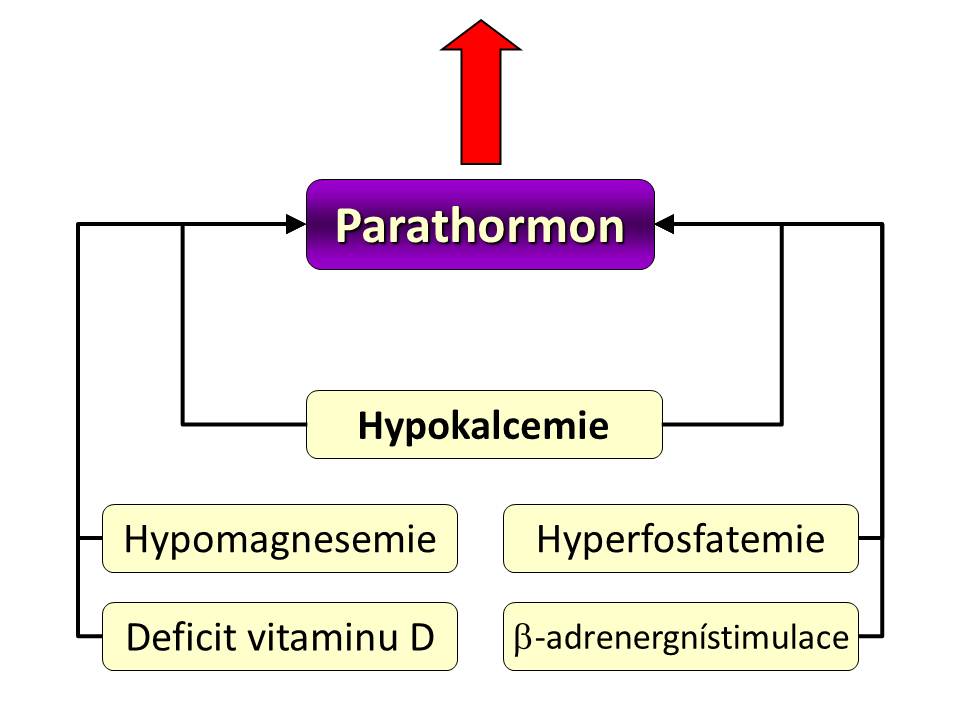 [1]
[1]Podněty zvyšující tvorbu a uvolňování PTH
- Hypokalcemie přesněji pokles hladiny ionizovaného vápníku plazmě představuje hlavní podnět pro zvýšení syntézy a sekrece parathormonu. Čidlem kalcemie resp. jeho koncentrace v celém extracelulárním prostoru je calcium sensing receptor (CaSR, viz níže) na povrchu buněk příštítných tělísek, jenž patří do rodiny receptorů spojených s G proteiny. Hyperkalcemie a tím i zvýšená vazba volných Ca2+ na CaSR, zvyšuje jeho aktivitu a to vede ke snížení výlevu PTH z příštítných tělísek, naopak hypokalcemie snižuje aktivitu CaSR, což odbrzdí tvorbu a sekreci PTH v hlavních buňkách. Pokud hypokalcemie trvá déle, vede to k podpoře růstu – hyperplazii příštítných tělísek.
- Hypomagnesemie. Magnesemie stejně jako kalcemie ovlivňuje negativně zpětnovazebně sekreci patarathormonu, i když síla tohoto signálu (= schopnost Mg vázat se a měnit aktivitu calcium sensing receptoru) buněk příštítných tělísek a tím měnit sekreci PTH je asi 2x – 3 x nižší než pro vápník. Hypomagnesemie tedy zvyšuje, kdežto hypermagnesemie snižuje sekreci PTH. V protikladu k tomuto ovšem chronická hypomagnesemie inhibuje syntézu PTH a současně vede k rezistenci cílových tkání, především kostí na účinek PTH.
- Hyperfosfatemie zvyšuje uvolňování PTH jednak přímo a jednak nepřímo recipročním snížením kalcemie. Chronická hyperfosfatemie, kterou vídáme při chronickém renálním selhání, vede k hyperplazii příštítných tělísek a sekundární hyperparathyreose.
- Snížená hladina kalcitriolu. 1,25-(OH)2D neboli kalcitriol vazbou na receptory pro vitamin D (VDR receptor) vytváří komplex, který se váže na vitamin D responzibilní oblast v jádře a tím tlumí transkripci genu pro PTH, zvyšuje počet CaSR receptorů a také inhibuje proliferaci parathyreoidálních buněk. Tudíž při hypovitaminóze D z jakýchkoliv příčin nebo při rezistenci příštítných tělísek na vitamin D (down-regulace VDR receptorů např. při chronickém renálním selhání vlivem hyperfosfatemie) se transkripce genu a tím i syntéza PTH zvyšuje.
- β-adrenergní stimulace či inhibitory fosfodiesterázy zvyšují sekreci PTH zvýšením intracelulární koncentrace cAMP
- α-adrenergní stimulace a prostaglandiny naopak inhibují sekreci PTH snížením cAMP v buňkách příštítných tělísek.
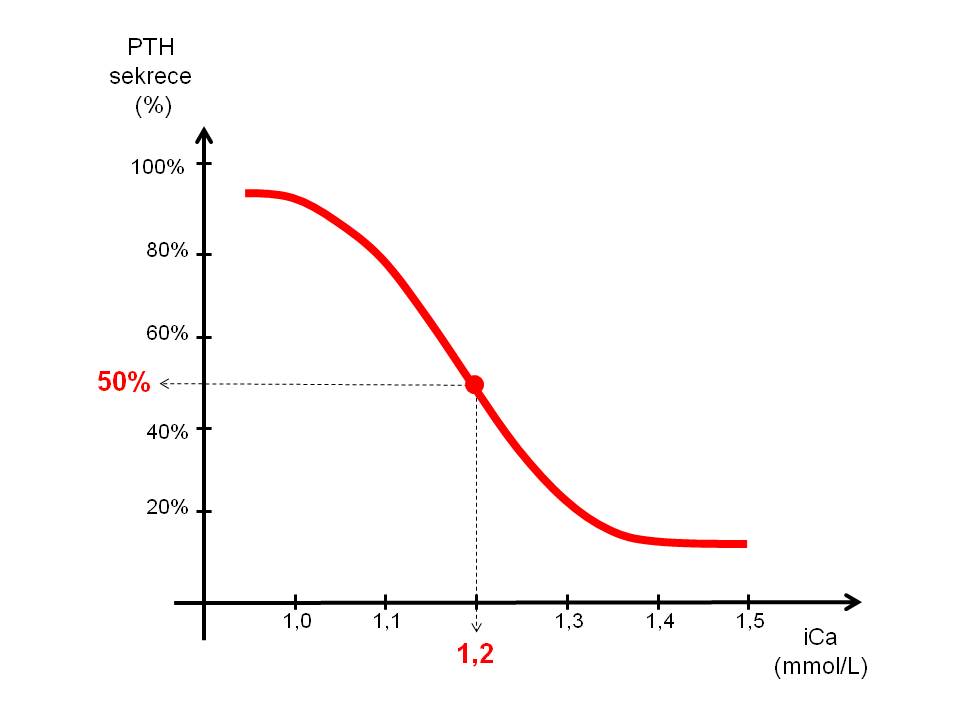
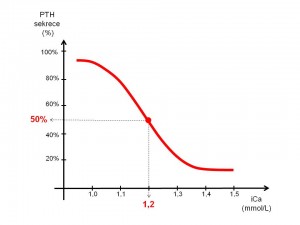 [2]
[2]Závislost sekrece PTH na koncentraci ionizovaného vápníku v plazmě
Calcium sensing receptor (CaSR)
- Extracelulární doménu vázající ligand, kterým mohou být nejen ionty vápníku či hořčíku, ale i jiných bivalentních iontů či dokonce aminokyseliny. Po navázání ligandu, v našem případě dostatečného množství iontů Ca2+ dojde ke konformační změně a tím i změně aktivity receptoru.
- Centrální doménu uloženou v membráně buněk a složenou ze 7 částí spojených extra a intracelulárními kličkami
- Intracelulární doménu směřující do cytoplazmy a schopnou vázat G proteiny a tím zajišťovat přenos signálu do nitra buňky.
- CaSR v příštítných tělíscích reguluje syntézu a sekreci parathormonu
- CaSR v ledvinách nacházíme v celém tubulárním systému. Nejvíce poznatků máme o jeho funkci v buňkách silných ramének Henleových kliček, kde je exprimován na bazolaterální membráně a reguluje zpětnou resorpci Ca2+ a Mg2+ kationtů, kdy zvýšená aktivita CaSR vede ke sníženému zpětnému vstřebávání vápníku a hořčíku, kdežto snížená aktivita CaSR vede ke zvýšenému zpětnému vstřebávání obou kationtů. Podstatou je změna transpeiteliálního elektrického gradientu, který představuje hnací sílu pro výstup bivalentních kationtů z lumina cestou paracelulárních zkratů. Ve vzestupné části Henleových kliček CaSR také reguluje zpětnou resorpci Na+ a Cl-. Zvýšená aktivita CaSR zde inhibuje resorpci soli přes natrium-kalium-chloridový kontransportér (NKKC2) a to pravděpodobně snížením aktivity draslíkového kanálu ROMK (renal outer medularry potassium channel), kterým draslík „recirkuluje“ zpět z tubulárních buněk do lumina, což je nezbytné pro činnost NKKC2. V proximálních tubulech ho nacházíme na apikální membráně, kde jeho aktivace zvyšuje zpětnou reabsorpci fosfátů v protikladu k účinku PTH a zřejmě zde i zvyšuje reabsorpci sodíku. V distálních tubulech a sběracích kanálcích je CaSR rovněž přítomen, jeho funkce zde zatím není příliš jasná, nicméně vliv na tubulární transporty bude jistě mít, už jen z předpokladu, že aktivace CaSR spojená s inhibicí adenylátcyklásy vede ke snížení produkce cAMP, který je 2.poslem účinku ADH a tímto mechanismem by (mimo účinku na Henleovu kličku) mohl CaSR přispívat k polyurii při hyperkalcemii. CaSR je i na bazolaterální membráně buněk macula densa a tím nepochybně (parakrinně) ovlivňuje sekreci reninu a tonus arteriola afferens.
- CaSR v gastrointestinálním traktu je prokázán od žaludku až po tlusté střevo, kde reguluje transport vody a iontů (Na+, Cl-, Ca2+, Mg2+). V žaludku se cestou CaSR zvyšuje sekrece HCl a to jednak přímo díky přítomnosti CaSR na bazocelulární membráně parientálních buněk a také nepřímo přes zvýšení sekrece gastrinu G-buňkami žaludku, a duodena jejichž membrána rovněž nese Ca sensing receptory. V tenkém a tlustém střevě se kromě regulace resorpce Ca2+ a Mg2+ podílí na regulaci sekrece/resorpce H2O a NaCl (aktivace CaSR spojená se snížením hladiny cAMP v enterocytech blokuje sekreci střevních šťáv). Navíc nedávné studie prokázaly vztah mezi zvýšeným dietním přívodem vápníku a sníženým rizikem rozvoje karcinomu tlustého střeva, což souvisí zřejmě s podporou diferenciace a inhibicí buněčného růstu epitelu tlustého střeva.
- CaSR v kardiovaskulárním systému nacházíme jak v srdci, tak v hladké svalovině cév, kde jeho aktivace vede hyperpolarizaci membrány myocytů (aktivace Ca2+ dependentního membránového kanálu pro K+ s následným přesunem draslíku vynášejícího pozitivní náboj z buňky ven) s následnou vazodilatací a poklesem krevního tlaku, což je podpořeno i uvolněním NO z endotelu cév, který na svém povrchu rovněž nese calcium sensing receptory. Z epidemiologických studií vyplívá, že zvýšený příjem Ca2+ v pitné vodě snižuje krevní tlak i dlouhodobě. Zde by vysvětlení prostým snížením periferní rezistence aktivací CaSR nestačilo, klíčem k dlouhodobé regulaci krevního tlaku jsou totiž ledviny a propojení regulace krevního tlaku a objemu tělesných tekutin. Vysvětlení by mohlo spočívat v modulaci tlakové diurézy vápníkem, protože CaSR jak již bylo uvedeno výše, nacházíme v celém tubulárním systému (přímé snížení zpětné reabsorpce sodíku do organismu při aktivaci CaSR) a také v juxtaglomerulárním aparátu (nepřímé snížení reabsorpce sodíku přes inhibici systému renin-angiotenzin II-aldosteron).
- CaSR v dalších orgánech a tkáních: Receptor je exprimován také v kostech, parafolikulárních buňkách štítné žlázy, což nepochybně souvisí s kalciumfosfátovým metabolismem, ale i v mozku, kůži, kostní dřeni, oční čočce, kde si musíme ještě počkat na objasnění jeho funkce.
Účinek parathormonu
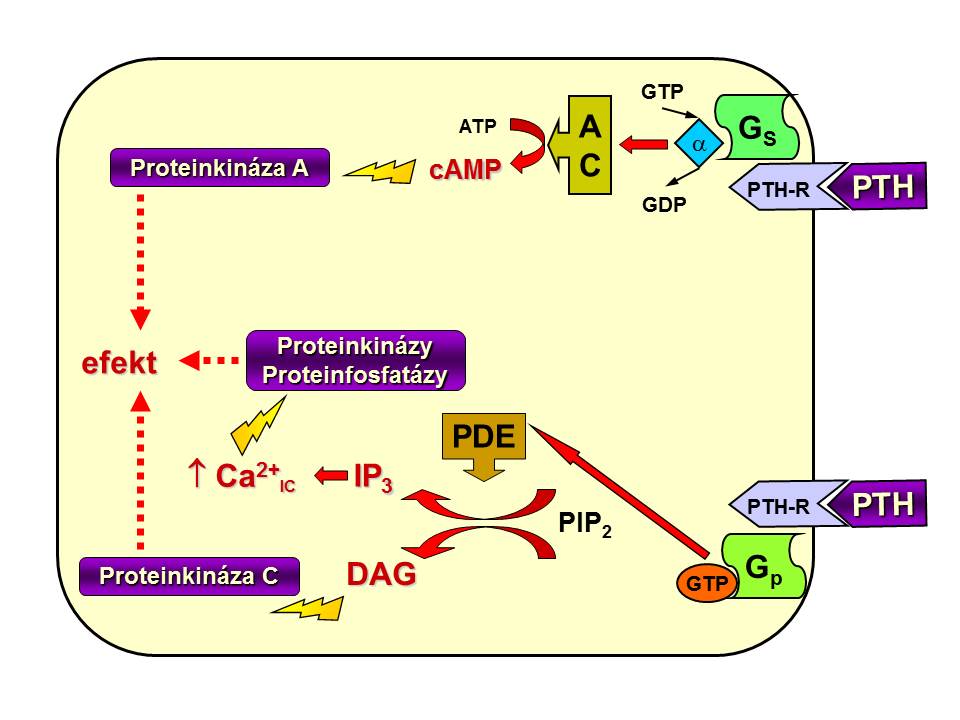 [3]
[3]PTH-receptor a jeho spřažení s G-proteiny
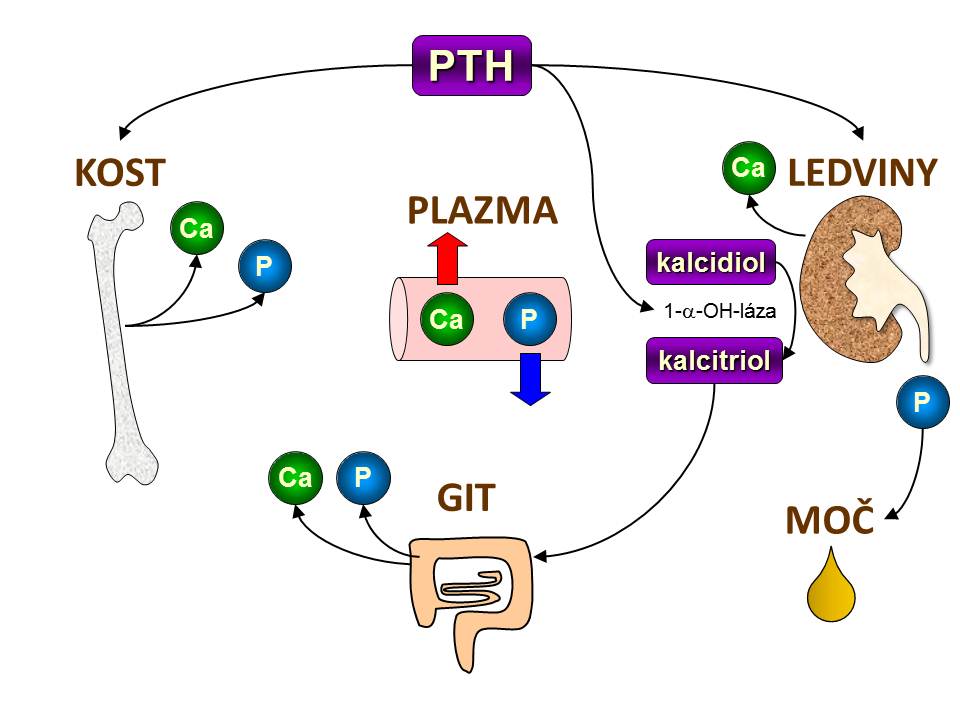 [4]
[4]Základní účinky PTH na kalciumfosfátový metabolismus
- Na PTHR1, který nacházíme v kostech i ledvinách, se mimo vlastního PTH může vázat i PTH related peptid (PTHrP), který se vytváří jak za fyziologických okolností (viz níže) tak i patologicky u některých typů nádorů. Vazba je zprostředkována N-terminální částí PTH mezi 1. – 34. aminokyselinou. Tento úsek představuje biologicky aktivní část PTH a vyskytuje se v plazmě i samostatně jako fragment PTH (1-34) vzniklý rozštěpením intaktního PTH (iPTH, PTH 1-84) v periferii, jak o tom bude pohovořeno v podkapitole o degradaci parathormonu (viz níže). Po aktivaci receptoru dojde k odštěpení podjednotek G proteinů, stimulaci adenylátcyklázy a fosfolipázy C, tvorbě 2. poslů v podobě cAMP a IP3, Ca2+ , fosforylaci cílových bílkovin s následnou změnou aktivity cílových tkání.
- PTHR2. Vazebnou sekvencí aktivující PTHR2 není na rozdíl od PTHR1 N-terminální část molekuly PTH, ale C-terminální část PTH či C-terminální fragment. Na receptor se není tudíž schopen vázat PTHrP. PTHR2 se kromě kostí vyskytuje v CNS, varlatech a pankreatu, jeho význam v těchto tkáních je nejasný.
PTH a kost
- Fáze aktivace prekurzorů osteoklastů z monocyt/makrofágové linie buněk hematopoézy v kostní dřeni a jejich maturace v mnohojaderné zralé osteoklasty.
- Fáze resorpce kosti osteoklasty, které těsně přilnou k povrchu kosti a produkcí agresivních působků, jako jsou H+ ionty, kyselá fosfatáza, katepsin K, rozrušují jak minerální složku tak, organickou složku kostní tkáně a vytváří resorpční dutinu (ve spongiózní kosti) či resorpční tunel (v kompaktní kosti).
- Fáze obratu, kdy osteoresorpce ustává, do vzniklé dutiny migrují preosteoblasty mezenchymálního původu. Tyto prekurzorové buňky proliferují a diferencují se vlivem růstových faktorů v osteoblasty.
- Fáze formace nové kosti. Osteoblasty vyplní resorpční kavitu osteoidem, organickou kostní matrix, jejíž hlavní složkou je kolagen I. typu, který se následně mineralizuje vápníkem a fosfáty. Po dokončení mineralizace zůstávají osteoblasty „uvězněny“ v kostní hmotě a transformují se v osteofyty.
- stimuluje činnost osteoblastů a novotvorbu kostí – účinek anabolický
- stimuluje aktivitu osteoklastů a odbourávání kostí – účinek katabolický
 [5]
[5]Regulace diferenciace osteoblastů - Signalizační cesta Wnt/beta-katenin
- zvyšuje přežívání preosteoblastů
- kontroluje genovou expresi transkripčních faktorů osteoblastů jako je Cbfa-1
- zvyšuje lokální tvorbu růstových faktorů jako IGF-1, TGFb
- inhibuje apoptózu osteoblastů.
- aktivuje Wnt/b-cateninovou cestu, která se podílí na proliferaci a diferenciaci osteoblastů, tuto cestu totiž inhibuje protein sklerostin produkovaný osteocyty a právě jeho produkci PTH blokuje.
Regulace diferenciace osteoklastů - Systém RANK/RANKL/OPG
PTH a ledviny
- Zvyšuje zpětnou tubulární resorpci vápníku, tím snižuje jeho exkreci z organismu, což představuje další cestu ke zvýšení hladiny plazmatického kalcia vedle již zmíněného uvolňování vápníku z kostí, které je rovněž pod kontrolou PTH. Místem účinku a následné zpětné resorpce Ca2+ je distální tubulus a tlustá část vzestupného raménka Henleovy kličky.
- Snižuje zpětnou tubulární resorpci fosfátů, tím zvyšuje jejich vylučování z těla. Podstatou účinku je snížení počtu Na+/fosfátových symportérů v luminální membráně buněk proximálního tubulu. Výsledná hyperfosfaturie a hypofosfatemie je ve zdánlivém paradoxu k účinku PTH na kosti spojeného se zvýšeným uvolňováním fosfátů, ale tento efekt je zcela nepostradatelný, protože současný vzestup kalcemie a fosfatemie by byl nežádoucí, mohlo by dojít k precipitaci fosforečňanu vápenatého a poškození orgánů.
- Zvyšuje konverzi kalcidiolu na kalcitriol a tím nepřímo zvyšuje vstřebávání vápníku a fosfátů ze zažívacího traktu – viz další podkapitola.
PTH a střevo
Degradace parathormonu
- N – terminální fragment, PTH 1-34 neboli teriparatid představuje vlastní biologicky aktivní část molekuly parathormonu vázající se na PTH receptory typu 1 s velmi krátkým biologickým poločasem 2 minut.
- C – terminální fragment, jeho poločas v plazmě je 30 -40 minut, filtruje se v glomerulech a je vylučován ledvinami. Byl považován za biologicky inaktivní část molekuly PTH, ale zdá se, že tomu tak není, váže se na druhý typ receptoru pro parathormon (PTHR2), v buněčných kulturách stimuluje syntézu ALP a dalších markerů osteoblastické aktivity, v jiných pokusech pro změnu zvyšoval apoptózu osteocytů.
Parathormonu podobný protein – PTH related peptid
- PTHrP kontroluje enchondrální osifikaci, tím že akceleruje růst chondrocytů, ale současně inhibuje jejich diferenciaci, umožňuje tak prodlužování kostí do délky z růstových chrupavek. Pohlavní hormony (androgeny, estrogeny) inhibují expresi PTHrP v chondrocytech a vedou proto k uzavření růstových štěrbin.
- PTHrP je zodpovědný za transfer vápníku od těhotné matky k plodu přes placentu, pravděpodobně tím, že stimuluje aktivní přesun vápníku přes placentu a to proti koncentrační gradientu, kalcemie plodu je vyšší než kalcemie u matky.
- PTHrP přispívá k mobilizaci vápníku z kostí u kojící ženy, která je potřebná pro zajištění dostatečné dodávky vápníku kojenému dítěti. Tvorba PTHrP probíhá v mléčné žláze a je kontrolována přes CaSR. PTHrP vstupuje nejen do cirkulace matky, ale dostává se i do mateřského mléka (jeho koncentrace zde je 104 oproti jeho koncentraci v plazmě) a může tak mít vliv na kalciumfosfátový metabolismus kojence.
- PTHrP se podílí na resorpci alveolární výběžku čelistních kostí, vývoji a prořezávání zubů
- PTHrP reguluje diferenciaci keratinocytů v kůži a růst vlasových folikulů
Hyperparathyreóza
Definice
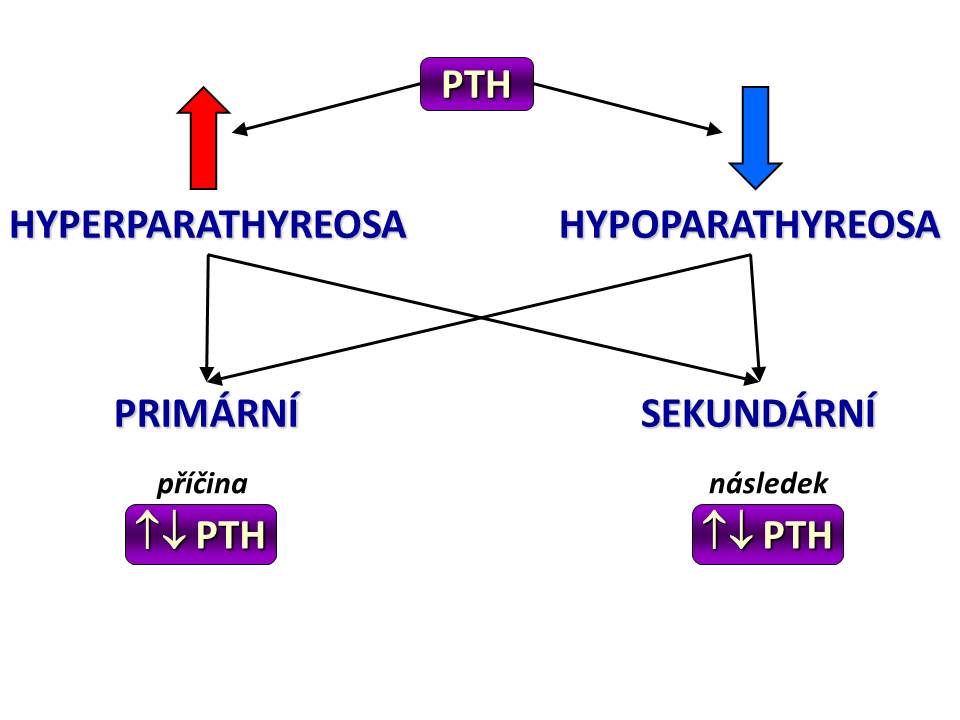 [7]
[7]Základní rozdělení poruch sekrece parathormonu
Primární hyperparathyreóza
Příčiny
- Solitární adenom příštítných tělísek, jde o nejčastější příčinu, 70 – 80 % všech primárních hyperparathyróz. Adenom se chová autonomně a secernuje PTH i přes hyperkalcemii.
- Idiopatická primární hyperplazie příštítných tělísek, ta se vyskytuje asi v 15 – 20 % případů, hyperplastická jsou všechna příštítná tělíska.
- Karcinom příštítných tělísek, který je velmi vzácný, je nalézán u 1 – 2 % jedinců s primární hyperparathyreózou, prognóza pacientů je špatná.
- Familiární hyperparathyreóza. Jde o genetická postižení vedoucí ke vzniku hormonálně aktivních nádorů nebo hyperplazie příštítných tělísek, patří sem
- Mnohočetné adenomy příštítných tělísek v rámci mnohočetné endokrinní neoplazie (MEN), příčina je genetická, výskyt familiární a na rozdíl od primární hyperparathyreózy při solitárního adenomu se onemocnění manifestuje v mladším věku. Z MEN syndromů hyperparatyreózu nacházíme v rámci MEN 1 a MEN 2a. MEN 1 je způsoben inaktivační mutací genu pro menin, který je součástí „tumor supresor genu“. Výsledkem mutace jsou kromě primární hyperparathyreózy na podkladě mnohopočetné adenomatózy také adenomy hypofýzy, které jsou buď afunkční (častěji) nebo hormonálně aktivní (nejčastěji prolaktinom), a do třetice nádory vycházející z buněk Langerhansových ostrůvků slinivky, z nichž nejčastějí je gastrinom, dále pak inzulinom, méně často glukagonom či VIPom. MEN 2a má příčinu v mutaci RET proto-onkogenu, gen kóduje receptor s tyrozinkinázovou aktivitou, mutací dojde k trvalé aktivaci tohoto receptoru. Klinickým následkem je u všech nemocných v průběhu jejich života dříve či později vznik medulárního karcinomu štítné žlázy (MTC), u asi poloviny pacientů se objeví feochromocytom a u jedné čtvrtiny primární hyperparathyreóza. K vývoji MTC dochází často už ve velmi mladém věku, proto je v případě průkazu této mutace u rodinných příslušníků pacienta s MTC doporučena preventivní totální thyreoidektomie v raném dětství.
- Familiární benigní hypokalciurická hyperkalcemie (FBHH), postižení jedinci jsou heterozygotní nositelé inaktivační mutace v genu pro calcium sensing receptor s autozomálně dominantním typem dědičnosti. Funkčním následkem je snížená citlivost příštítných tělísek na výši kalcemie, takže k útlumu sekrece parathormonu dochází až při vyšších hladinách plazmatické koncentrace vápníku neboli normální hladiny kalcia jsou příštitnými tělísky vnímány jako nízké. Klinicky jsou nositelé v naprosté většině případů asymptomatičtí a k podrobnějšímu diagnostickému vede až náhodný nález mírné hyperkalcemie, která je provázena hypokalciurií a hladinou PTH při horní hranici normy, ale nepřiměřenou k aktuální kalcemii. Léčba FBHH není potřebná k postižení kostí či ledvin nedochází.
- Těžká novorozenecká primární hyperparathyreóza se manifestuje u homozygotních nositelů inaktivační mutace pro CaSR, život ohrožující hyperkalcemie se objevuje už v novorozeneckém věku, hladiny PTH jsou extrémně vysoké. Již v nitroděložní fázi vývoje začíná a následně během prvních měsíců života se rozvíjí velmi těžká generalizovaná demineralizace skeletu vedoucí k mnohočetným frakturám. Měkký poddajný deformovaný hrudník v kombinaci se svalovou hypotonií může vést k respiračnímu selhání. Děti neprospívají, špatně tolerují příjem stravy, zvrací, mají zácpu a pokud přežijí první rok života tak jen se závažnou psychomotorickou retardací. Jedinou kauzální terapií je u nich ablace všech příštítných tělísek s následnou suplementací kalcia a vitaminu D.
Neuromuskulární projevy
Renální projevy primární hyperparathyreózy
Kostní projevy
Gastrointestinální projevy
Kardiovaskulární projevy
Neuropsychiatrické projevy
Kloubní, oční, kožní projevy
Laboratorní nálezy
- vysoká plazmatická koncentrace iPTH – prvotní příčina problému
- hyperkalcemie – výsledek zvýšeného uvolňování vápníku z kostí při převaze aktivity osteoklastů nad osteoblasty, zvýšené zpětná resorpce Ca2+ v ledvinných tubulech a nepřímo zvýšené střevní resorpce
- hypofosfatemie – i přes zvýšené uvolňování fosfátů z kostí, protože PTH blokuje resorpci fosfátů v proximálních tubulech ledvin
- hyperkalciurie – na první pohled překvapivý nález, protože PTH zvyšuje tubulární reabsorpci Ca2+, ale nálož vápníku, která je v glomerulárním filtrátu přesahuje kapacitu zpětné resorpce a proto jsou odpady kalcia močí vysoké
- hyperfosfaturie – viz hypofosfatemie
- zvýšená hladina 1,25-(OH)2-D je dána jeho vystupňovanou tvorbou v ledvinách vlivem vysoké hladiny PTH a následně zvyšuje kalcemii zvýšeným vstřebáváním Ca2+ v ledvinách
- zvýšená alkalická fosfatáza. ALP respektive její kostní izoforma je markerem kostní novotvorby, aktivity osteoblastů a proto je zvýšená také u hyperparathyreózy, která je provázena zvýšeným kostním obratem
- zvýšená kyselá fosfatáza. ACP je markerem kostní resorpce a primární hyperparathyreóza je charakteristická výrazně vystupňovanou aktivitou osteoklastů (zvýšena je aktivita osteoblastů i osteklastů, ale osteoklasty dominují)
- hyperchloremická metabolická acidóza. PTH snižuje reabsorpci bikarbonátu v proximálmních tubulech, chybějící bikarbonát, kromě toho že chybí jako pufr, je pak v rámci zachování elektroneutrality nahrazen chloridovým aniontem, který je ovšem na rozdíl od bikarbonátu aniontem silné kyseliny a pH plazmy se proto snižuje.
Sekundární hyperparathyreosa
Příčiny
- Renální insuficience resp. renální selhání, které jsou provázeny komplexní poruchou kalciumfosfátového metabolismu. Hlavní patogenetickou příčinou je hyperfosfatemie, která je výsledkem stupňující se retence fosfátů při poklesu glomerulární filtrace (hyperfosfatemie se objevuje při poklesu GFR pod 0,5 ml/s, do té doby nestoupá, protože vlivem PTH se zvyšuje exkreční frakce fosfátů, která dokáže kompenzovat snižující se glomerulární filtraci fosfátů). Hyperfosfatemie (a) přímo stimuluje tvorbu PTH v příštítných tělíscích, (b) vede k rezistenci příštítných tělísek na kalcitriol, který naopak sekreci PTH snižuje, rezistence je dána sníženým počtem receptorů pro vitamin D (VDR), (c) snižuje aktivitu 1-a-hydroxylázy v ledvinách a tím samotnou tvorbu kalcitriolu. Nicméně k deficitu kalcitriolu dochází ještě před objevením se hyperfosfatemie a to v souvislosti s úbytkem funkčního renálního parenchymu (hypokalcitriolemie se objevuje při poklesu GFR na hodnoty pod 1,0 ml/s, před tím je pokles celkové kapacity enzymu kompenzován zvýšením jeho aktivity vlivem vysoké hladiny PTH). Deficit kalcitriolu (absolutní daný jeho sníženou produkcí a současně relativní při rezistenci příštítných tělísek na jeho účine) vede ke sníženému vstřebávání vápníku ze střeva, což společně s rezistencí kostí na PTH, která je pravděpodobně rovněž dána hyperfosfatemií, podporuje vznik hypokalcemie. K hypokalcemii přispívá samotná hyperfosfatemie, protože jak bylo řečeno v úvodu tohoto příspěvku, vztah mezi kalcemií a fosfatemií je reciproční, tak aby nedošlo k jejich vzájemné precipitaci. Trias hyperfosfatemie, deficit kalcitriolu a hypokalcemie zvyšuje tvorbu a sekreci PTH a při delším trvání vede k hyperplazii všech příštítných tělísek. Poločas účinku PTH je navíc prodloužen pro jeho sníženou renální clearenci.Výsledkem je sekundární hyperparathyreosa a renální osteodystrofie, která zahrnuje jak změny vídané při hyperparathyreózní osteodystrofii tak při osteomalacii. Pacienti i přes hypokalcemii nemívají křeče, což je dáno současně přítomnou metabolickou acidózou, která zvyšuje podíl ionizované frakce vápníku na celkové kalcemii, současně metabolická acidosa podporuje kostní resorpci. Sekundární hyperparathyreosa je v rámci renální insuficience mechanismus adaptační, který má normalizovat plazmatické hladiny vápníku a fosfátů, ale ve chvíli, kdy i přes maximální zvýšení exkreční frakce fosfátů dochází k hromadění fosfátů v organismu a vzestupu jejich plazmatické koncentrace, začíná být zvýšená hladina PTH kontraproduktivní, protože vystupňovaná kostní resorpce dále zvyšuje hladinu fosfátů bez možnosti současné renální korekce, což vede k devastaci kostí a současně ke vzniku extraoseálních kalcifikací, například v cévách.
- Hypovitaminóza D. Vitamin D získáváme jednak z potravy a to jak živočisné (mléčné výrobky, ryby) v podobě cholekalciferolu tak i rostlinné (obiloviny, zelenina) v podobě ergokalciferolu. Obě formy jsou rozpustné v tucích a proto jejich vstřebání závisí na absorpci tuků v GIT. Druhým a obvykle dominantním (90 %) zdrojem vitaminu D je naše kůže, kde vzniká cholekalciferol syntézou de novo z cholesterolu vlivem slunečního záření. Nedostatek vitaminu D v lidském těle tak vzniká (a) nedostatkem vitaminu D ve stravě (typicky kojené děti, protože mateřské mléko ač jinak dokonalé ve svém složení neobsahuje dostatek tohoto vitaminu, (b) nedostatečným vstřebáváním vitaminu D z GIT v rámci malabsorpčních syndromů (jako je celiakie, insuficience exikrinního pankreatu či tvorby žluči atd.) a současně s jednou nebo druhou možností je přítomna (c) nedostatečná tvorba vitaminu D v kůži (nedostatečný sluneční osvit v zimních mědsících, u malých dětí, u jedinců tmavé pleti, přiliš „dokonalé“ opalovací krémy). Nedostatek vitaminu D pak vede k nedostatečnému vstřebávání vápníku (ale i fosfátů) ze střeva a vzniklý deficit v bilanci Ca2+ vede k hypokalcemii, která stimuluje tvorbu a vylučování parathormonu. Postižení kostí následkem hypovitaminózy D probíhá u dětí v podobě rachitidy, u dospělých v podobě osteomalacie. V širším slova smyslu můžeme do příčin hypovitaminózy D zahrnout i (d) stavy snížené aktivace vitaminu D v organismu (vitamin D prochází dvojí hydroxylací, první v játrech a druhou v ledvinách, která jeho účinnost zvyšuje o tři řády) v rámci jaterního selhání či renálního selhání a (e) antagonizaci jeho účinku tak jak to dělají endogenní či exogenní glukokortikoidy
- Malabsorpční syndromy, které postihují vstřebávání vápníku, sem patří již zmiňovaná celiakie, poruchy sekrece žluči a pankreatické šťávy a jsou spojeny s malabsorpcí tuků. Snížená resorpce Ca2+ je zapříčeněna dvěmi skutečnostmi: (a) tvorbou vápenatých mýdel, která vznikají reakcí mezi ionty vápníku a mastnými kyselinami, (b) nedostatečným vstřebáváním vitaminu D, který je závislý na vstřebání tuků.
Laboratorní nálezy u sekundární hyperparathyreózy
- vysoká plazmatická koncentrace iPTH – stejně jako u primární hyperparathyreosy, ale zde jde o změnu sekundární
- hypo/normokalcemie – zvýšená hladina parathormonu má za úkol kompenzovat primárně sníženou hladinu vápníku
- hyper/hypofosfatemie – hladina fosfátů závisí na příčině sekundární hyperparathyreózy, při renální etiologii je přítomna hyperfosfatemie, u deficititu vitaminu D je obvykle hypofosfatemie
- hyperkalciurie – na první pohled překvapivý nález, protože PTH zvyšuje tubulární reabsorpci Ca2+, ale nálož vápníku, která je v glomerulárním filtrátu přesahuje kapacitu zpětné resorpce a proto jsou odpady kalcia močí vysoké.
- hyperfosfaturie – viz hypofosfatemie
- snížená hladina kalcitriolu je typická pro renální formu sekundární hyperparathyreózy, kde se v důsledku úbytku funkční ledvinné tkáně snižuje kapacita a současně vlivem hyperfosfatemie i aktivita 1a-hydroxylázy a tím i tvorba kalcitriolu.
- zvýšené markery kostního obratu: zvýšená ALP, ACP a další.
- zvýšená plazmatická koncentrace močoviny (P-U) a kreatininu (P-Kr) jako důsledek poklesu renálních forem pomáhají v diferenciální diagnostice sekundární hyperparathyreózy.
Hypoparathyreóza
Definice
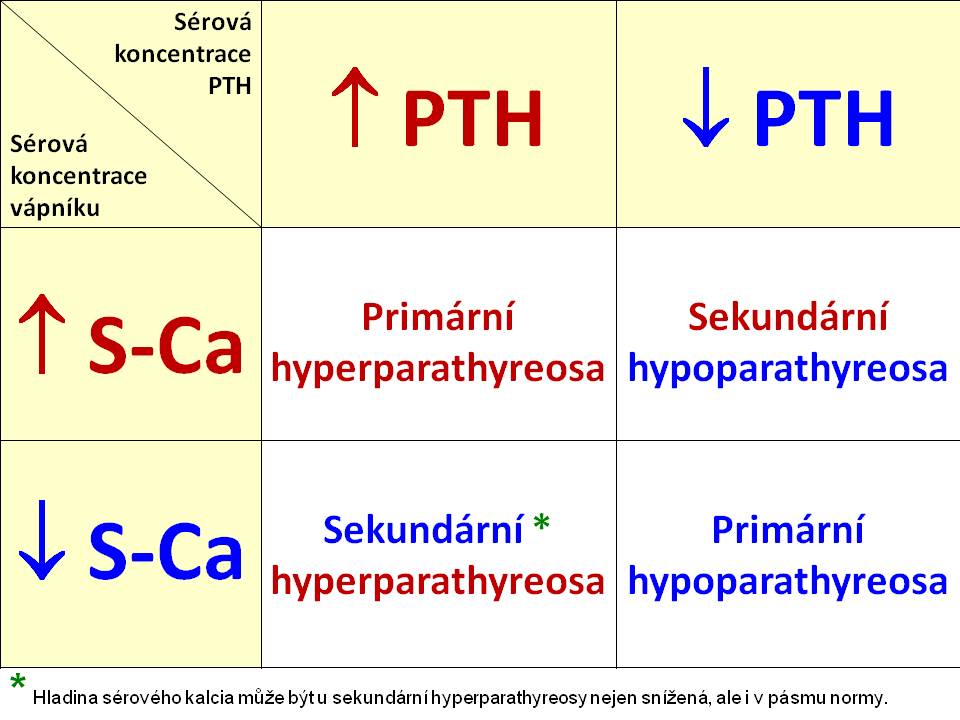 [8]
[8]Základní diferenciace poruch sekrece PTH na podkladě hladiny vápníku a PTH v plazmě
Primární hypoparathyreóza
Příčiny primární hypoparathyreózy
Stavy vedoucí k primární hypoparathyreóze můžeme rozdělit na získané a vrozené, přičemž první jmenované jsou mnohem častější.
- Poškození příštítných tělísek v souvislosti s operacemi na štítné žláze je nejčastější příčinou primární hypoparathyreózy. Snahou operatéra provádějícího thyreidektomii je snaha zachovat alespoň jedno příštítné tělísko, ale ne vždy se to zdaří a příštítná tělíska tak mohou být všechna odstraněna společně se štítnicí. Někdy hypoparathyreóza vzniká, i když 1 tělísko zůstane zachováno, může být ovšem postiženo peroperační hypoxií nebo destruováno pooperační fibrotizací v lůžku po štítné žláze. Z klinického pohledu se rozlišuje přechodná hypoparathyreóza jejíž trvání je omezeno na dny až týdny a hypothyreosa trvalá, která vyžaduje doživotní léčbu.
- Poškození příštítných tělísek radiací může být komplikací léčby radiojodem u hyperfunkčních strum či hyperfunkčních adenomů štítnice nebo jako součást terapie při karcinomu štítné žlázy, dále při zevním ozařování krku u nádorů v ORL oblasti.
- Poškození příštítných tělísek u metabolických chorob jako je Wilsonova choroba (defekt v metabolismu mědi) či hemochromatóza (defekt v metabolismu železa), kdy dochází k pozitivní bilanci Cu či Fe v organismu s jejich ukládáním v různých tkáních vč. příštítných tělísek, oba tyto prvky se mohou vyskytovat ve dvojmocné (Fe2+, Cu2+) či trojmocné podobě (Fe3+ , Cu3+) a mohou se tak stát donory elektronů v tzv. Fentonově reakci, která vede ke vzniku kyslíkových radikálů a ty potom poškozují tkáně v nichž jsou zmiňované tranzientní kovy naakumulovány.
- Autoimunní hypoparathyreosa. Stejně jako jiné endokrinní žlázy mohou se i příštítná tělíska státi terčem autoagresivní reakce imunitního systému, který vede k postupnému poklesu jejich funkce. V tomto případě je hypoparathyreóza obvykle součástí autoimunního polyglandulárního syndromu typu I (APS I), který je autosomálně recesivně dědičným onemocněním. Příčinou je mutace genu AIRE (autoimunne regulator), který je lokalizován na dlouhém raménku 21. chromozomu. Pro klinické určení této diagnosy je třeba přítomnosti dvou ze tří hlavních projevů a to je: chronická mukokutánní kandidóza (bývá prvním příznakem, objevuje se do 5 let věku dítěte), primární hypoparathyreosa (je nejčastějším endokrinním projevem APS-1 a většinou se objeví do 10 let věku, primární hypokorticismus (Addisonova choroba se obvykle projeví do 15. roku života a představuje hlavní příčinu předčasné smrti pacientů s APS-1). Pacientni s APS-1 ovšem mohou být postiženi i dalšími endokrinními autoimunitami (AI hypotyreóza, DM 1. typu, hypergonadotropní hypogonadismus) , ale i neendokrinními autoimunitami (alopecie, vitiligo, percinózní anemie, atrofická gastritida, AI hepatitida, ulcerozní kolitida a další).
- Kongenitální familiární hypoparathyreóza patří k vzácným příčinám, dědí se různým způsobem (AD, AR, X-vázaná dědičnost) a projevuje se izolovanou familiární hypoparathyreosou.
- DiGeorgův syndrom je porucha vývoje orgánů jejichž základy pochází ze 3. a 4. žaberního oblouku a vzniká v důsledku mikrodelece a translokace na 22. chromozomu. Syndrom v sobě zahrnuje aplázii příštítných tělísek, která je příčinou hypoparathyreosy, aplazii thymu, která vede k imunodeficienci a vrozené vývojové vady srdce (Fallotova tetralogie) a aorty (interrupce aortálního oblouku, truncus arteriosus), které se mohou projevit příznaky srdečního selhání. V neposlední řadě u postižených DiGeorgovým syndromem bývají přítomna fenotypická stigmata v obličeji (mikrognacie, šikmo posazené oční stěrbiny, nízce posazené a tvarově deformované ušní boltce, rozštěp patra).
- Tranzientní novorozenecká hypoparathyreosa může být přechodným problémem u dítěte, jehož matka měla v průběhu těhotenství hyperparathyreosu, což vedlo k útlumu funkce příštítných tělísek plodu.
- Funkční hypoparathyreóza po odstranění hyperfunkčního adenomu příštítných tělísek je rovněž přechodným problémem a to u jedinců jimž byl vyoperován hormonálně aktivní adenom příštítných tělísek. Ten po dobu své existence vedl k dlouhotrvající hyperkalcemii, která utlumila nepostižený zdravý parenchym příštítných tělísek. Po jeho odstranění vzniká hypokalcemie z nedostatečné sekrece PTH potencována zvýšeným vychytáváním kalcia kostmi po odstranění tumoru (syndrom hladové kosti)
Neuromuskulární příznaky
- Chvostkův příznak. Při klepnutí do oblasti průběhu n.facialis před ušním boltcem dojde ke stažení ústního koutku, nosního křídla a očního koutku.
- Lustův příznak. Poklepem na průběh n.peroneus pod hlavičkou fibuly dojde k dorzoflexi a abdukci nohy.
- Trousseaův příznak. Vyvolání karpálního spazmu při stažení paže manžetou tonometru na úroveň systolického tlaku po dobu cca 3 minut.
- Erbův příznak. Dráždění periferního nervu stejnosměrným proudem vede ke vzniku tetanického stahu příslušné svalové skupiny již při malé intenzitě (pod 5 mA) tohoto proudu.
- Mezi první/prodromální příznaky patří parestézie prstů, rtů, jazyka, které jsou projevem zvýšené excitability periferních senzitivních nervů. Dále mohou být pozorovány fascikulace (drobné jemné záškuby svalových snopců), postižený si stěžuje na pocit tuhnutí/napnutí svalů.
- Pokud vezmeme v úvahu kosterní svalovinu, pak vlastní tetanický záchvat nejčastěji probíháve formě karpo/pedálních spazmů: Na horních končetinách se ruce stahují do extenze, palec je adduktován do dlaně, zápěstí je ve flexi, postavení je přirovnáváno k „porodnické ruce“, křeč se může šířit proximálně s flexí v lokti a addukcí v rameni. Na dolních končetinách jde noha do plantární flexe, koleno je v extenzi. Tetanické stahy jsou značně bolestivé, při těžší hypokalcemii dochází k jejich generalizaci, obvykle začínají na akrech a směřují proximálně, jsou bolestivé, na rozdíl od epileptických záchvatů zůstává pacient při plném vědomí! Nejnebezpečnější je postižení svalů hrtanu a svalů dýchacích, postižený jedinec má inspirační stridor, cyanosu, dusí se. Tento laryngosmazmus a spazmus respiračních svalů může skončit fatálně.
- Zvýšenou excitabilitu vykazuje i hladká svalovina a to se může projevit jako bronchospazmus s expiračním stridorem, dysfagie pro spazmus jícnového svěrače, křeče v břiše pro stahy hladké svaloviny střev, žlučníkovou kolikou atp.
- Hypokalcemie logicky ovlivňuje i elektrické děje na myokardu. Na EKG nacházíme prodloužení ST úseku a tím i QT intervalu, které je dáno prodloužením fáze platau akčního potenciálu na komorách. Pravděpodobné vysvětlení je obdobné, ale opačné než jak tomu bylo při hyperkalcemii v rámci hyperparathyreosy. Při hypokalcermii je chemický gradient pro vstup Ca2+ menší a tak je prodloužena doba po kterou jsou otevřeny vápníkové kanály, kterými Ca2+ proudí do kardiomyocytů. Nedostatek vápníku uvnitř pracovního myokardu pak snižuje jeho inotropii, což může vést poruše kontraktility srdce a ve finále k srdečnímu selhání.
Neuropsychiatrické příznaky
Kostní a zubní příznaky primární hypoparathyreosy
Kožní a oční příznaky
Laboratorní nálezy u primární hypoparathyreózy
- nízká plazmatická koncentrace iPTH – prvotní příčina chorobného stavu
- hypokalcemie – nedostatek PTH vede k negativní bilanci kalcia (snížené vstřebávání Ca2+ v GIT, zvýšená exkrece ledvinami), která nemůže být ani vykompenzována uvolněním Ca2+ z kostí, jde tedy o základní laboratorní nález a současně o příčinu zvýšené nervosvalové dráždivosti
- hyperfosfatemie – z nedostatečné renální exkrece fosfátů při chybění PTH
- hypokalciurie – chybění PTH sice snižuje tubulární reabsorpci Ca2+, ale nálož vápníku, která je v glomerulárním filtrátu je malá a tak zvýšené vylučování Ca2+ nevzniká, naopak exkrece je snížená
- hypofosfaturie – viz hypofosfatemie
- snížená hladina kalcitriolu je následkem snížené aktivity 1a-hydroxylázy v ledvinách
Sekundární hyporathyreóza
- Deficit hořčíku/hypomagnesemie. Akutní hypomagnesemie sice zvyšuje sekreci PTH, ale chronický deficit Mg snižuje sekreci parathormonu a současně snižuje odpověď kostí na účinek PTH (rezistence), protože nedostatek hořčíku snižuje mobilizaci vápníku z kostí, jelikož vápník nahrazuje magnesium, které se z kostí uvolňuje, aby doplnilo hladinu Mg v plazmě a v ostatních orgánech
- Hypervitaminóza D. Nadbytek vitaminu D vede ke zvýšení hladiny jeho aktivního metabolitu kalcitriolu a ten snižuje syntézu a sekreci parathormonu, která je současně tlumena i hyperkalcemií vznikající následkem zvýšené střevní resorpce Ca2+ vlivem účinku vitaminu D na enterocyty. Navíc vysoké dávky vitaminu D vedou ke zvýšenému uvolňování kalcia z kostí a zhoršení hyperkalcemie.
- Zvýšená sekrece PTHrP. O PTH podobnému peptidu, příčinách jeho zvýšené tvorby a následcích byla řeč výše. Funkčně jde o hyperparathyreózu s hyperkalcemickým syndromem, ale hladina samotného PTH je nízká, tlumená vysokou koncentrací plazmatického vápníku a proto bývá někdy tento paraneoplastický syndrom zaškatulkován mezi sekundární hypoparathyreózy, lepší název by ale byl pseudohyperparathyreosa analogicky k faktu, že existuje pseudohypoparathyreosa, které se budeme věnovat na následujících řádcích.
Pseudohypoparathyreóza
- Pseudohypoparathyreosa typ Ia neboli Albrightova hereditární osteodystrofie je způsobena maternálně zděděnou autosomálně dominantní inaktivační mutací a podjednotky Gs proteinu kódované genem GNAS1. Součástí tohoto syndromu jsou somatické kostní anomalie: nanismus, kulatý obličej, široký nos, krátký krk, krátké končetiny, brachydaktylie (zkrácené jsou zejména 4. a 5. prsty). Současně je porušena účinnost TSH a gonadotropinů LH, FSH s následnou sekundární hypothyreózou a sekundárním hypogonadismem.
- Pseudohypoparathyreosa typ Ib. Genetický defekt je způsoben ztrátou metylace v jednom z exonů genu GNAS1 což souvisí s mikrodelecí v genu pro syntaxin 16, který se nachází v jeho těsném sousedství. Rezistence postihuje jen účinek PTH v ledvinách, na rozdíl od typu Ia proto chybí výše uvedené klinické anomalie, diagnosa je laboratorní s nálezem hypokalcemie, hyperfosfatemie při vysoké hladině PTH a normální glomerulární filtrací (normální P-U, P-Kr).
- Pseudohyhypoparathyreóza typ II. Genetický defekt je někde na postreceptorové úrovni, somatické anomalie chybí, laboratoř odpovídá pseudohypoparathyreóze, odlišení od typu I b je možné exogením podáním PTH, kdy u typu II dojde ke zvýšení exkreci cAMP do moči, kdežto u typu Ib ke zvýšení nedojde.
- Pseudo-pseudohypoparathyreóza je způsobena paternálně přenesenou inaktivační mutací genu GNAS1. Postižení jedinci mají skeletální anomalie Albrightova typu, ale laboratorní parametry kalciumfosfátového metabolismu jsou v normě.
Literární zdroje
- BERNE RB, LEVY MN, KOEPPEN BM, STANTON BA. Physiology, 5th edition, Mosby 2004, pp. 22 – 30, 806 – 813
- GEIBEL JP, HEBERT SC. The functions and role sof the exttracellular Ca2+ sensing receptor along the gastrointestinal tract. Annu.Rev.Physiol. 2009 71: 205-17
- GUYTON AC, HALL JE. Textbook of medical physiology, 11th edition, Elsevier Saunders 2006, pp. 985 – 988
- JABOR a kol. Vnitřní prostředí. 1.vydání, Grada 2008, str. 211-215
- KHOSLA S., Minireview: The OPG/RANKL/RANK Systém. Endocrinology 2001, 142(12): 5050-5055
- KOVACS CS, KRONENBERG HM. Maternal-fetal calcium and bone metabolism during pregnancy, puerpium and lactation.Endocrine Rewiews, 1997, 18(6): 832-872
- MISIOROWSKI W. Parathyroid hormone and its analogues – molecular mechanisms of action and efficacy in osteoporosis therapy. Polish J. of Endocr. 2011, 62(1): 73-78
- MUNDY GR, EDWARDS JR. PTH-related peptide in hypercalcemia.J Am Coc Nephrol, 2008, 19: 672-675
- OBRMANOVÁ B, ŠUMNÍK Z, CINEK O, LEBL J. Kalcium sensing receptor: Fyziologie a onemocnění spojená s jeho poruchami, DMEV 2009, 4(12): 193 – 200
- POOL K, REEVE J. Parathyroid hormone – a bone anabolic and catabolic agent. Current opinion in Pharmacology, 2005, 5: 612-617
- SLÁMA O. Hyperkalcemie u maligních onemocnění. Paliativna medicín a liečba bolesti, 2009, 2(2): 84-85
- Van HOUTEN J, DANN P, McGEOCH G, BROWN EM, KRAPCHO K, NEVILLE M and WYSOLMERSKI JJ. The calcium-sensing receptor regulates mammary gland parathyroid hormone – related protein production and calcium transport. J Clin Investigation, 2004,113(4): 598-608
Article printed from Tvorba a ověření e-learningového prostředí pro integraci výuky preklinických a klinických předmětů na LF a FZV UP Olomouc: http://pfyziolklin.upol.cz
URL to article: http://pfyziolklin.upol.cz/?p=6346
URLs in this post:
[1] Image: http://pfyziolklin.upol.cz/wp-content/uploads/2012/09/PTH-stimuly-sekrece.jpg
[2] Image: http://pfyziolklin.upol.cz/wp-content/uploads/2013/04/PTH-vztah-sekrece-a-iCa.jpg
[3] Image: http://pfyziolklin.upol.cz/wp-content/uploads/2012/09/PTH-R-a-efekt-na-bunku.jpg
[4] Image: http://pfyziolklin.upol.cz/wp-content/uploads/2012/09/PTH-zakladni-ucinky.jpg
[5] Image: http://pfyziolklin.upol.cz/wp-content/uploads/2012/09/PTH-a-diferenciace-osteoblastu.jpg
[6] Image: http://pfyziolklin.upol.cz/wp-content/uploads/2012/09/PTH-a-system-RANK-RANKL-OPG.jpg
[7] Image: http://pfyziolklin.upol.cz/wp-content/uploads/2012/09/PTH-deleni-poruch.jpg
[8] Image: http://pfyziolklin.upol.cz/wp-content/uploads/2012/09/PTH-tabulka-poruch.jpg
Click here to print.