Téma: Sodný iont, hyponatremické a hypernatremické stavy
Posted By vodouch On 16.3.2012 @ 13:47 In 5.2. Poruchy iontové rovnováhy | Comments Disabled
pracoviště: Ústav patologické fyziologie LF UP Olomouc, Dětská endokrinologická ambulance Svitavské nemocnice a.s.
Úvod
Sodík neboli natrium (11Na, Ar = 23,0) je nenahraditelným biogenním prvkem, který zastává řadu fyziologických funkcí. Společně s dalšími ionty a soluty díky své osmotické aktivitě udržuje vodu v těle . Do našeho organismu se ionty sodíku dostávají především jako stolní sůl neboli chlorid sodný (NaCl).
Pradávní předkové člověka opustili moře, a tím i zdroj soli, ve kterém bylo snadné udržet Na+ homeostázu. Suchozemským tvorům nezbylo než fylogeneticky vyřešit nedostatek soli tím, že se u nich vyvinuly velmi silné natrium-retenční mechanismy, které jsou schopné zajistit vyrovnanou bilanci sodíku i při velmi nízkém přísunu soli do organismu.
Sůl byla ještě v nedávné historii významnou, vysoce ceněnou obchodní a strategickou surovinou. Ovšem v současnosti, díky cenové dostupnosti soli a přemíře jejího využití v potravinářském průmyslu, se naše sodík-spořící geny obrací proti nám a výsledkem je vzestup výskytu arteriální hypertenze a s ní spojených dalších civilizačních onemocnění. Z pohádky „Sůl nad zlato“ se tak pro naši populaci stává horor.
Zastoupení iontů sodíku v organismu
 [1]
[1]Distribuce sodíku v lidském organismu
- 10 % sodných iontů je uloženo intracelulárně, tzn. uvnitř buněk, jeho koncentrace v tomto prostoru kolísá mezi 3 – 35 mmol/l s průměrem okolo 12 – 15 mmol/l.
- 50 % sodných iontů se nachází v extracelulární tekutině, což u dospělého člověka vážícího 70 – 80 kg představuje asi 2000 mmol Na+. Sodík je kvantitativně nejvýznamnějším extracelulárním kationtem a extracelulární tekutina je největší zásobárnou Na+. Extracelulární koncentrace sodíku je rovna plazmatické koncentraci sodíku neboli natrémii, která je fyziologicky udržována okolo 140 ± 5 mmol/l. Naprostá většina sodíku v plazmě (99 %) je ve formě volného, ionizovaného Na+, zbývající 1 % je vázáno v komplexech nebo na plazmatické bílkoviny.
- 40 % sodných iontů je vázáno v kostech a dalších tkáních. Tato frakce sodíku má pomalý obrat, a tudíž nemá význam při akutních výkyvech v bilanci sodíku.
Příjem iontů sodíku ve stravě
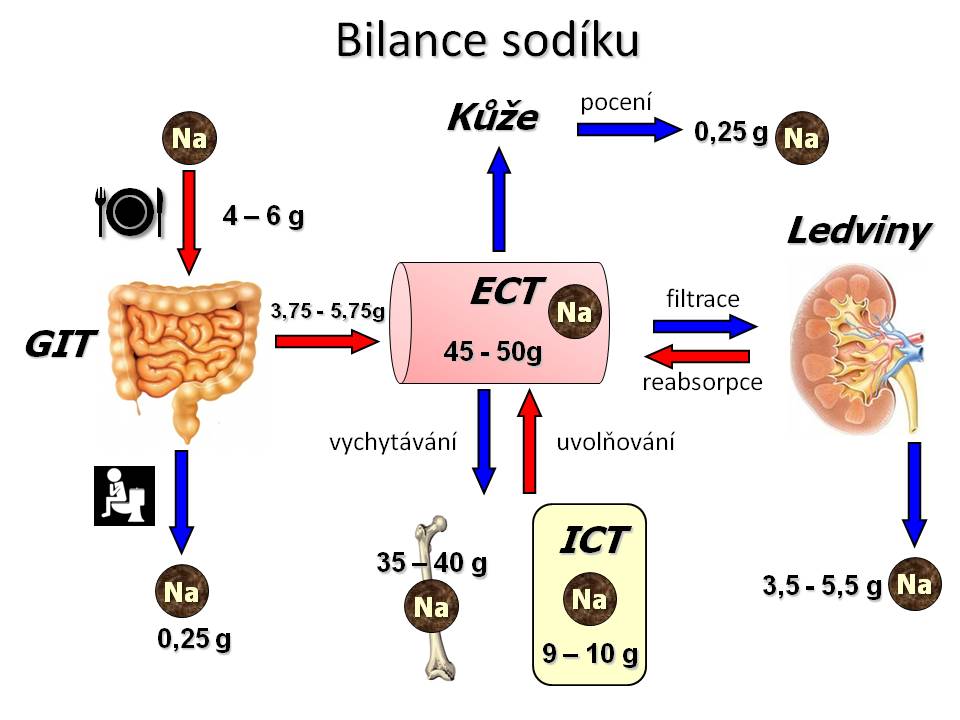 [2]
[2]Denní bilance sodíku
Vstřebání a vylučování iontů sodíku v GIT
Distribuce iontů sodíku mezi ECT a ICT
- dostatku ATP;
- dostatku substrátu, tedy Na+ uvnitř buněk a K+ vně buněk;
- dostatku hořčíku, protože ionty Mg2+ jsou součástí aktivního místa molekuly Na+/K+-ATPázy.
Vylučování iontů sodíku močí
- FENa = [(U-Na x S-Kr)/(S-Na x U-Kr)] x 100,
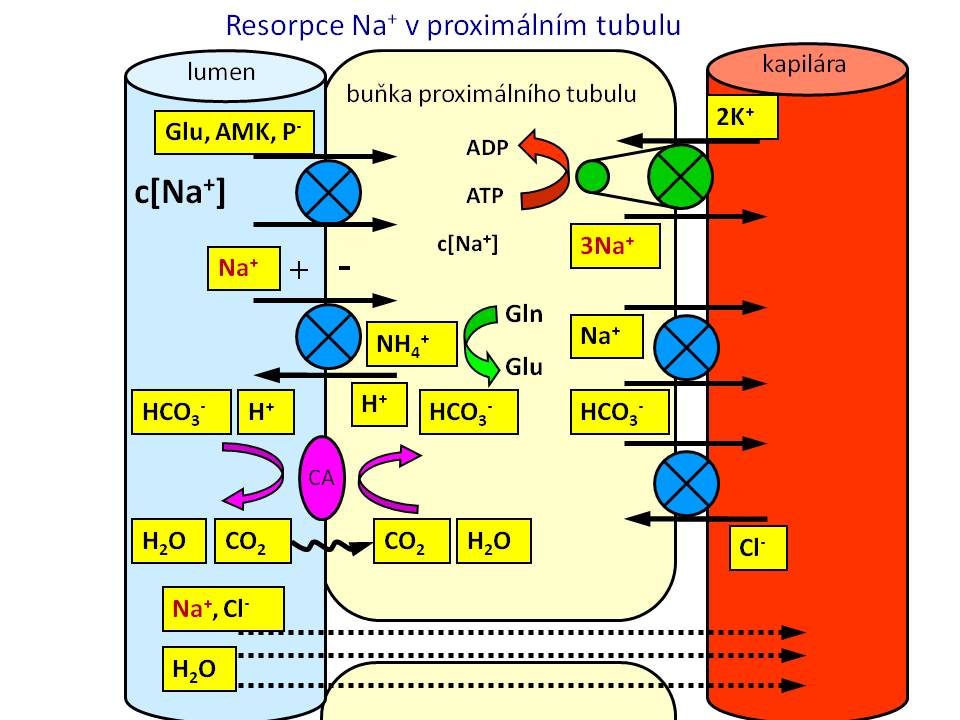 [3]
[3]Resorpce sodíku v proximálním tubulu
Proximální tubulus
Zde se vstřebává největší část, a to 65 % iontů sodíku profiltrovaných v glomerulech. Mechanismus resorpce je jak aktivní, tak pasivní.
- Aktivní resorpce je zajištěna činností Na+/K+-ATPázy umístěné na bazolaterální membráně tubulární buňky, která vypuzením iontů sodíku ven z buňky do perikapilárního prostoru vytváří příznivý elektrochemický gradient (nízká koncentrace Na+ uvnitř buněk společně s negativním nábojem) pro resorpci sodných iontů z tubulární tekutiny přes luminální membránu do nitra tubulární buňky. Přes luminální membránu se sodné ionty dostávají prostřednictvím různých transportních proteinů. Po funkční stránce jde buď o sekundárně aktivní kotransport nebo protitransport, kdy je resorpce sodíku po příznivém gradientu spojena s resorpcí, nebo naopak sekrecí, další látky, která přestupuje luminální membránu proti svému (elektro)chemickému gradientu. Příkladem budiž kotransport Na+-glukóza anebo Na+-aminokyselina nebo protitransport Na+/H+.
- Pasivní resorpce je dána fyzikálním jevem zvaným tah rozpustidla. Proximální tubulus je totiž vysoce propustný pro vodu, přičemž voda se pohybuje za solí po osmotickém spádu vytvořeném aktivní resorpcí Na+. Tento tok či „tah“ vody s sebou strhává přes paracelulární štěrbiny další ionty a soluty včetně iontů sodíku. Poměr resorpce Na+ a vody proto v proximálním tubulu je 1 : 1, a díky tomu je tekutina odtékající z proximálního tubulu do Henleovy kličky izoosmolární.
-
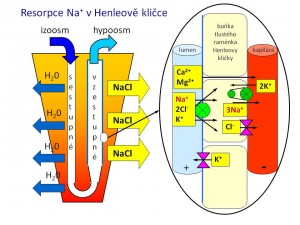 [4]
[4]Resorpce sodíku v Henleově kličce
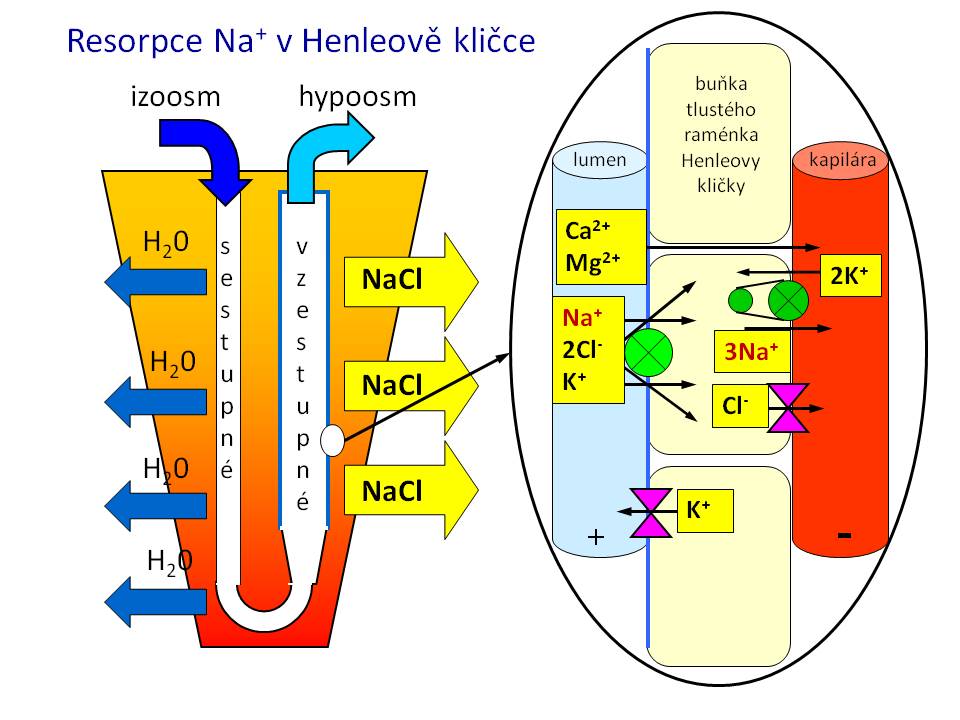
Henleova klička
Tenká část Henleovy kličky je propustná pro vodu, které se zde resorbuje asi 20 %, ale není prostupná pro sůl. K resorpci Na+ proto dochází až ve vzestupné tlusté části, a to v množství odpovídajícím 25 % profiltrovaných sodných iontů. Stejně jako v proximálním tubulu je i zde na bazolaterální straně membrány přítomna Na+/K+-pumpa, která za spotřeby ATP vytváří resorpční gradient. Ionty sodíku se zde resorbují společně s ionty draslíku a s chloridy přes NKCC2 (Na-K-Cl-cotransporter). Tento kanál je možné farmakologicky zablokovat tzv. kličkovými diuretiky, jako je například furosemid. Na celkovém množství sodných iontů vstřebaných v Henleově kličce má významný podíl i pasivní resorpce přes paracelulární zkraty. Její hnací silou je relativně pozitivní intraluminální potenciál oproti intersticiu, který z lumina „vytlačuje“ pozitivně nabité kationty včetně kationtů sodíku. Tekutina opouštějící Henleovu kličku je hypoosmolární, protože resorpce iontů v této části převažuje nad resorpcí vody.
Distální tubulus
Zde se zpětně resorbuje asi 5 % profiltrovaných iontů sodíku, přičemž na luminální membráně je vstup sodných iontů do tubulárních buněk zajištěn přítomností transportního proteinu NCC (Na-Cl-cotransporter) citlivého na thiazidy (jde o skupinu účinných diuretik). Na bazolaterální membráně pak jsou ionty sodíku vypuzovány z buněk do intersticia stejně jako v ostatních částech nefronu, tedy aktivní činností Na+/K+-ATPáz.
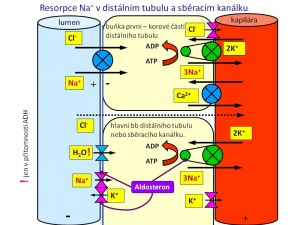 [5]
[5]- Resorpce sodíku v distálním tubulu a sběracím kanálku
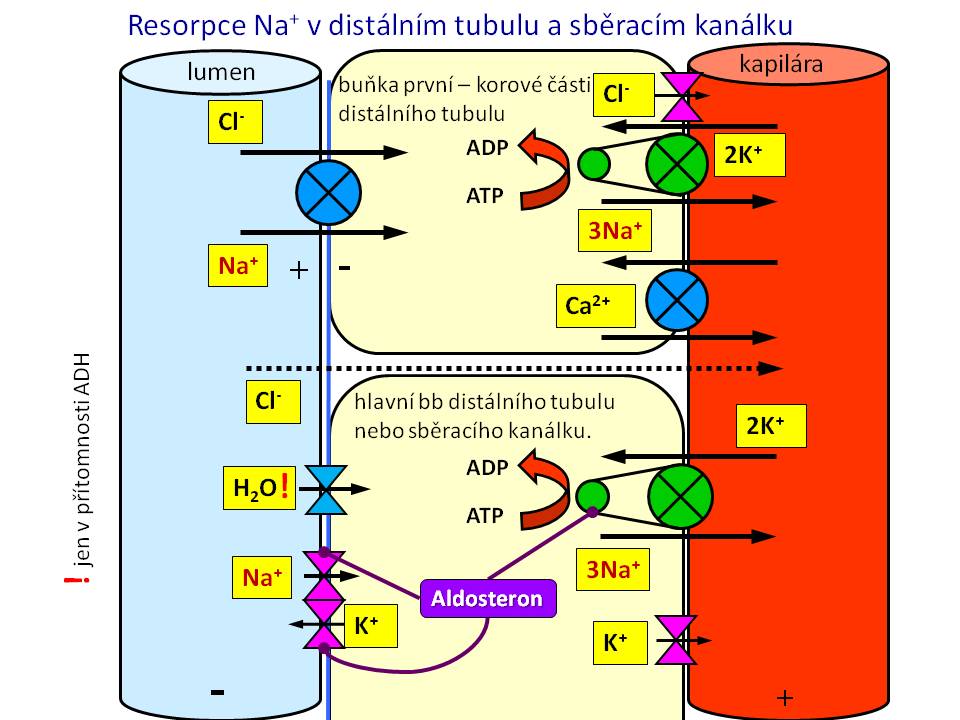
Sběrací kanálek
Tato část (funkčně zahrnuje i konečnou část distálního tubulu) odpovídá za finální úpravu množství sodných iontů v definitivní moči dle potřeb organismu. Obvykle se zde resorbují asi 4 % profiltrovaných iontů sodíku. Zpětná resorpce iontů sodíku je zde pod hormonální kontrolou aldosteronu, který stimuluje jeho zpětné vstřebání. Na apikální membráně hlavních buněk sběracích kanálků probíhá resorpce Na+ za současné sekrece K+. Na+ přestupují z lumina do buněk přes epiteliální sodíkový kanál (ENaC), kdežto K+ unikají z buněk do lumina tubulů přes ROMK kanály. Na této úrovni působí tzv. kalium-šetřící diuretika, kam patří jednak antagonisté aldosteronu (spironolakton), jednak blokátory kanálu ENaC (amilorid, triamteren).
- Glomerulotubulární rovnováha (GTB). Pokud se organismus nachází ve stavu vyrovnané bilance iontů sodíku, pak zvýšení glomerulární filtrace, a tím zvýšené množství filtrovaného Na+, vede ke zvýšení zpětné tubulární reabsorpce sodíku v proximálních tubulech, a naopak snížení glomerulární filtrace, a tím i filtrace Na+, vede ke snížení zpětné reabsorpce sodíku v proximálních tubulech. Jinými slovy, navzdory výkyvům glomerulární filtrace je z proximálního tubulu resorbována zpět konstantní frakce sodných iontů a vody. Podstatou GTB jsou reciproční změny Starlingových kapilárních sil vyvolané změnou GFR. Uvažované síly působí mezi peritubulárním prostorem (tj. intersticiem okolo tubulárních buněk) a peritubulární sítí kapilár. Jedná se o čtyři známé Starlingovy síly - hydrostatický tlak v peritubulárních kapilárách (Pc), onkotický tlak v těchto kapilárách (Πc), hydrostatický tlak v peritubulárním intersticiu (Pi) a onkotický tlak v peritubulárním intersticiu (Πi). Zatímco Pi a Πc podporují vstřebávání tekutiny z peritubulárního prostoru do kapilár, a tím i resorpci sodných iontů v proximálním tubulu, působí Pc a Πi proti vstřebávání tekutiny z peritubulárního prostoru do kapiláry. Tím snižují resorpci iontů sodíku v proximálním tubulu. Rozdíl těchto sil pak určuje tzv. efektivní filtrační tlak (Pef), kde Pef = [Pi + Πc] – [Pc + Πi]. Za fyziologických okolností platí, že Pi + Πc > Pc + Πi, tudíž je favorizován pohyb vody, iontů a solutů z intercelulárního prostoru mezi tubulárními buňkami směrem do kapilár. Současně je zajištěna stabilní resorpční frakce ultrafiltrátu v proximálním tubulu (činí okolo 2/3 profiltrovaného množství). Proč? Jestliže dojde ke snížení GFR, méně krve se profiltruje, tudíž vzroste hydrostatický tlak v peritubulárních kapilárách (Pc), současně však zůstanou více „naředěny“ plazmatické bílkoviny, které glomerulárním filtrem neprostupují, a to vede k poklesu onkotického tlaku v kapilárách (Πc). Výsledný efektivní filtrační tlak klesá ([Pi + ↓ Πc] – [↑ Pc + Πi] = ↓↓Pef), a proto klesá i síla ženoucí tekutinu z intersticia do kapilár. Dochází k úniku vody a solutů přes těsná spojení tubulárních buněk zpět do lumina proximálních tubulů, a tím se snižuje celková míra resorpce vody, sodných iontů i dalších iontů a solutů. Při zvýšení GFR je situace opačná - [Pi + ↑ Πc] – [↓ Pc a Πi] = ↑↑ Pef. Starlingovy síly nemají efekt na tlustou část Henleovy kličky, distální tubuly ani sběrací kanálky, protože tyto oddíly nefronu jsou jen málo propustné pro vodu. V případě distálního nefronu pak je permeabilita pro vodu řízena hormonálně prostřednictvím ADH.
- Tubuloglomerulární zpětná vazba (TGF). Při zvýšení GFR se zvýší přísun iontů sodíku k buňkám macula densa. Jde o specializované buňky na začátku stočené části distálního tubulu, které těsně přiléhají ke glomerulárním arteriolám a s jejich myoepiteliálními buňkami vytváří tzv. juxtaglomerulární komplex. Při zvýšeném přísunu iontů k macula densa se z jejích buněk zvýšeně uvolňuje ATP a molekuly ATP se pak vážou na purinergní receptory ve stěně a. afferens (buď přímo nebo nepřímo – po konverzi na adenosin). Výsledkem je vazokonstrikce a. afferens s následným poklesem filtrace iontů sodíku v glomerulech a snížením přítoku soli do oblasti macula densa. Tím se regulační okruh uzavírá. Úkolem TGF je vyrovnávat oscilace GFR.
Fyziologická regulace vylučování iontů sodíku ledvinami
Precizní řízení natriurézy je nezbytným předpokladem pro udržení vyrovnané homeostázy iontů sodíku, které je nutné pro přežití organismu. Fylogeneticky se proto vyvinula celá řada hormonálních i nehormonálních faktorů regulujících tubulární resorpci iontů sodíku. Z funkčního hlediska si tyto faktory můžeme rozdělit na natriumretenční a natriuretické.
Mechanismy a systémy natriumretenční
Mezi faktory, které zvyšují zpětnou resorpci iontů sodíku, a tím snižují natriurézu, patří:
 [6]
[6]- Systém renin-angiotenzin II-aldosteron

- Systém renin-angiotenzin (RAS). Renin je enzym (aminopeptidáza) vytvářený granulárními buňkami juxtaglomerulárního aparátu v ledvinách. Podněty pro tvorbu reninu jsou tři: (a) Snížený přítok NaCl tubulární tekutinou do oblasti macula densa jako následek snížené glomerulární filtrace. (b) Zvýšená aktivita sympatiku, která zvyšuje sekreci reninu přes β1 receptory umístěné na povrchu granulárních buněk. (c) Pokles perfúzního tlaku v ledvinách. Juxtaglomerulární buňky patří mezi baroreceptory, tj. jsou citlivé na změny krevního tlaku. Angiotenzinogen je plazmatický alfa-2-globulin syntetizovaný játry a slouží jako substrát pro renin. Angiotenzin I je oligopeptid, který vniká zkrácením N-terminálního konce angiotenzinogenu účinkem reninu. Angiotenzin II pak je vlastním účinným peptidovým hormonem systému RAS, který vzniká z angiotenzinu I účinkem angiotenzin-konvertujícího enzymu (ACE). Tvorba angiotenzinu II probíhá jak na úrovni systémové, a to především v plicích (dáno přítomností ACE), tak na úrovni ledvin v endotelu renálních cév, přičemž renálně dosahují jeho koncentrace mnohonásobně vyšších hodnot než v systémové cirkulaci. Angiotenzin II zvyšuje reabsorpci sodných iontů
- přímo přes AT-receptory v proximálním tubulu;
- nepřímo zvýšením tvorby aldosteronu;
- přes vazokonstrikci a. efferens, která snižuje tlak ve třeni ledvin a usnadňuje přestup iontů a vody z tubulů do intersticia.
 [7]
[7]Účinek aldosteronu v ledvinách
- Aldosteron je mineralokortikoid kůry nadledvin (její zona glomerulosa). Nejvýznamnějšími podněty pro jeho tvorbu a sekreci jsou angiotenzin II a hyperkalémie, naopak minoritní význam za fyziologických okolností má ACTH. Aldosteron zvyšuje zpětnou resorpci iontů sodíku hlavními buňkami distálního nefronu, a to následujícími mechanismy:
- Zvyšuje kapacitu systému Na+/K+-ATPáz v bazolaterální membráně (zvyšuje jejich počet);
- Zvyšuje permeabilitu apikální membrány pro ionty sodíku, protože zvyšuje počet ENaC kanálů, kterými sodné ionty proudí po elektrochemickém spádu z lumina tubulů do nitra hlavních buněk. Zvýšená resorpce iontů sodíku také je hnací silou zvýšené sekrece iontů draslíku, které přestupují přes ROMK kanály z nitra hlavních buněk do lumina tubulů. Rozvinutí účinků aldosteronu vyžaduje změnu proteosyntézy, a proto se dostavují s časovým zpožděním několika hodin.
- Sympatické autonomní nervstvo rovněž zvyšuje zpětnou reabsorpci iontů sodíku, a to od proximálním tubulu přes tlustou část Henleovy kličky a distální tubulus až po sběrací kanálky; ve výsledku zvyšuje efektivní cirkulující objem. Mechanismus, kterým tak sympatikus činí, je trojí:
- Zvyšuje sekreci reninu z granulárních buněk juxtaglomerulárního aparátu přes β1 receptory, a tím zvyšuje aktivitu systému renin-angiotenzin II-aldosteron (RAA).
- Vede k vazokonstrikci a. afferens přes α1-receptory, a tím snižuje hydrostatický tlak v glomerulárnm klubíčku, což má za následek snížení glomerulární filtrace.
- Přímo zvyšuje reabsorpci iontů sodíku tubulárními buňkami přes α2-receptory.

Mechanismy a systémy natriuretické
Mezi faktory, které snižují zpětnou resorpci iontů sodíku, a tím zvyšují natriurézu, patří:
 [8]
[8]Základní princip tlakové diurézy/natriurézy
- Tlaková diuréza-natriuréza. Představuje kruciální fyziologický mechanismus regulace krevního tlaku (viz obrázek). Ledviny udržují krevní tlak tím, že přizpůsobují objem krve v cirkulaci kapacitě krevního řečiště. Současně platí, že bilance tekutin v organismu je vyhodnocována pomocí změny krevního tlaku. Nemáme žádné receptory, které by přímo měřily objem tělesných tekutin anebo absolutní náplň řečistě, ale pouze baroreceptory, které monitorují relativní náplň řečiště, tj. objem ku kapacitě, tzn. krevní tlak. Při zvýšení arteriálního krevního tlaku a perfúzního tlaku v ledvinách dochází okamžitě (během 30 – 60 vteřin) k nárůstu natriurézy a diurézy a tento vzestup trvá do doby, než se krevní tlak vrátí k normě. Naopak pokles arteriálního a perfúzního tlaku v ledvinách vede ke snížení natriurézy a diurézy. Grafickým vyjádřením tlakové diurézy je tzv. funkční renální křivka, z jejíhož normálního strmého průběhu je patrné, že i malá změna krevního tlaku vede k velké změně natriurézy a diurézy (viz obrázek) .
 [9]
[9]Tlaková diuréza
K tomuto dochází i přes známý fakt, že průtok krve glomeruly a peritubulární oblastí je i při značných výkyvech středního arteriálního tlaku udržován na konstantní úrovni (GFR zůstává stabilní při kolísání MAP v rozpětí 70 – 160 mm Hg). Z toho plyne, že za změnu exkrece iontů Na+ pravděpodobně nejsou odpovědné změny filtrace sodíku v glomerulech, ale především změny reabsorpce Na+ v průběhu tubulů. Přesný mechanismus tlakové diurézy není znám, za hlavního kandidáta na mediátora tlakové natriurézy je považován oxid dusnatý (NO). Představa o jeho úloze je následující: Zvýšení perfúzního tlaku v ledvinách zvyšuje smykové tření, což v endotelu renálních cév a především a. afferens indukuje expresi syntázy oxidu dusnatého typu 3 (NOS 3); vzniklý NO difunduje do krve a přes glomerulární filtr do tubulů, kde parakrinním způsobem zvýší tvorbu 2. posla v podobě cGMP, který inhibuje zpětnou reabsorpci sodíku tubulárními buňkami, a tím zvyšuje natriurézu. Krevní tlak se tak stává nástrojem ledvin pro udržení vyrovnané bilance tekutin. Pokud v organismu dojde k poruše rovnováhy příjmu a výdeje vody a soli, bude krevní tlak obětován na normalizaci bilance, protože nerovnováha mezi příjmem a výdejem tekutin vede rychle ke smrti organismu, kdežto změna výše krevního tlaku je dlouhodobě slučitelná se životem.
- Atriální natriuretický faktor (ANF) je peptidický hormon tvořený atriálními kardiomyocyty. Stimulem je zvýšené napětí stěny srdečních síní vlivem zvýšeného plnění síní při zvýšeném efektivním cirkulujícím objemu anebo i při fyzickém cvičení. Tvorbu a uvolnění ANF rovněž podporuje β1-adrenergní stimulace, fibrilace síní nebo endotelin. V ledvinách ANF zvyšuje vylučování jak Na+, tak vody několika způsoby:
- Dilatací a. afferens a konstrikcí a. efferens zvyšuje glomerulární filtraci, a tím i finální množství moči a obsah soli v ní.
- Snižuje reabsorpci iontů sodíku a vody ve stočené části distálních tubulů a v kortikálních sběracích kanálcích vlivem cGMP-dependentní fosforylace ENaC.
- Snižuje sekreci reninu a aldosteronu.
- Zvýšením průtoku krve přes vasa recta snižuje obsahu iontů solutů v dřeni ledvin, tím klesá koncentrační schopnost a tubulární reabsorpce tekutin.
- Dopamin na rozdíl od ostatních katecholaminů působí pro-natriureticky, a to inhibicí zpětné reabsorpce NaCl v proximálním tubulu. Stimulem pro uvolnění dopaminu je zvýšení efektivního cirkulujícího objemu. Dopamin se v ledvinách jednak uvolňuje z dopaminergních neuronů (což je pro natriurézu nevýznamné) a jednak je tvořen buňkami proximálních tubulů. Syntéza dopaminu v proximálních tubulech není závislá na sympatiku. Substrátem pro syntézu je L-DOPA z glomerulárního ultrafiltrátu, jejíž přestup přes apikální membránu zajišťuje kotransportér stimulovaný sodnými ionty. Enzymem zajišťujícím syntézu je DOPA-dekarboxyláza, jejíž aktivita roste při zvýšeném přísunu soli. Nově syntetizovaný dopamin je pak vylučován do lumen tubulů a dostává se i do dalších etáží nefronu. Efekt dopaminu v ledvinných tubulech je zprostředkován především přes D1 receptory pro které je 2. poslem cAMP, ale část účinku je zprostředkována i přes další dopaminergní receptory (D2-D5). Dopamin zvyšuje natriurézu několika způsoby:
- Snižuje aktivitu Na+/K+-ATPázy na bazolaterální membráně, a to ve všech typech tubulů, takže méně sodných iontů se přesunuje směrem do peritubulárního prostoru. To snižuje příznivý koncentrační gradient pro přestup iontů sodíku z lumina do tubulárních buněk po celé délce tubulů.
- Snižuje aktivitu Na+/H+ výměníku a Na+/Px--kontransportéru v luminální membráně buněk proximálních tubulů, kterými sodné ionty vstupují do tubulární buňky (prostředníkem je změna produkce cAMP).
- Snižuje aktivitu Na+/3HCO3- symportéru na bazolaterální membráně proximálních tubulů, což snižuje příznivý elektrochemický gradient pro resorpci iontů sodíku v této části nefronu.
- Inhibuje sekreci a antagonizuje účinek RAS, aldosteronu a ADH.
- Urodilatin je peptidový hormon velmi blízký ANF (je delší o 4 aminokyseliny). Tvoří se jen v ledvinách v distálních tubulech a sběracích kanálcích a má jen renální účinky, v systémové cirkulaci ho nenajdeme. Podnětem pro jeho tvorbu je opět zvýšení efektivního cirkulujícího objemu. Natriuretický účinek urodilatinu spočívá v inhibici zpětné reabsorpce Na+ v medulární části sběracích kanálků. Účinek je obdobně jako u ANF zprostředkován přes cAMP. Urodilatin je ve srovnání s ANF mnohem účinnější, protože ANF je po vstupu do ledvin degradován endopeptidázou.
- Guanylin a uroguanylin jsou peptidové hormony produkované střevním epitelem a působící na epitelové buňky ve střevě, v tubulech ledvin, v dýchacích cestách a ve vývodných cestách orgánů (játra, slinivka) cestou receptorů spřažených s guanylát-cyklázou anebo G-proteiny (odtud jméno). Tvoří páteř osy střevo-ledviny. Stimulují exkreci sodných iontů i draselných iontů (následky zvýšené produkce jsou např. průjem anebo zvýšená natriuréza).

Fyziologický význam sodných iontů v organismu
Sodné ionty jsou pro život zcela nepostradatelné. Jejich fyziologické uplatnění si můžeme rozdělit na následující okruhy:
Na+ a tonicita a objem tělesných tekutin
Na+ a elektrická aktivita vzrušivých tkání
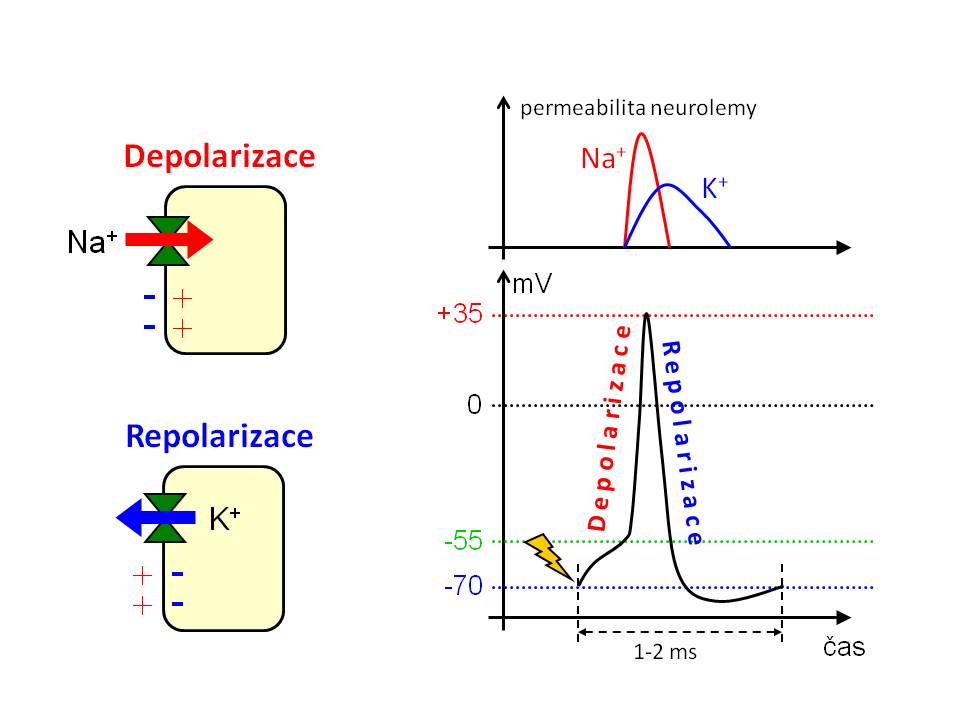 [10]
[10]Účast sodíku na průběhu akčního potenciálu neuronu
Na+ a acidobazická rovnováha
Na+ a sekundárně aktivní transporty
Definice hyponatrémie
Výskyt hyponatrémie
- vyšší věk,
- cukrovka,
- chronická ledvinná onemocnění,
- léčba diuretiky, opiáty, analgetiky,
- antibiotiky, plicní infekce,
- terapie hypotonickými roztoky,
- chirurgický zákrok aj.
Klasifikace hyponatremických stavů
 [11]
[11]Diferenciální diagnostika hyponatremií
Hypovolemická hyponatrémie
Při hypovolemické hyponatrémii se snížená plazmatická koncentrace Na+ kombinuje se sníženým objemem ECT. Vyšetřením pacienta nacházíme klinické anebo laboratorní známky dehydratace. Pacient má oschlé sliznice, povleklý jazyk, snížený kožní turgor, tachykardii, hypotenzi se sklonem ke kolapsům, snížený tonus očních bulbů.
Laboratorně bývá zvýšená plazmatická hladina močoviny (P-U), kreatininu (P-Kr), kyseliny močové (P-KM) a poměru P-U/P-Kr.
Obecnou příčinou tohoto typu hyponatrémie je ztráta hypernatremické tekutiny, tzn. ztráty sodíku jsou větší než ztráty vody. Výsledkem je depleční hyponatrémie. Teoretickou příčinou je snížený přívod Na+ do organismu, ale vzhledem k obsahu NaCl ve středoevropské stravě to opravdu nehrozí.
Euvolemická hyponatrémie
Při euvolemické hyponatrémii se snížená plazmatická koncentrace Na+ kombinuje s téměř normálním objemem ECT. Vyšetřením pacienta nenacházíme klinicky zjevné známky hyperhydratace ani dehydratace. Ve skutečnosti je u tohoto typu hyponatrémie objem ECT lehce zvýšen, což je prokazatelné až podrobnějším vyšetřením. Obecnou příčinou tohoto typu hyponatrémie je nadbytek čisté vody při normálním stavu zásob Na+ v organismu – jedná se tedy o čistou diluční hyponatrémii.
Hypervolemická hyponatrémie
Při hypervolemické hyponatrémii se snížená plazmatická koncentrace Na+ kombinuje se zvýšeným objemem ECT. Vyšetřením pacienta nacházíme klinické a laboratorní známky hyperhydratace. Pacient má podkožní otoky, může mít ascites, hydrothorax, hydroperikard, otok plic s dušností. Obecnou příčinou tohoto typu hyponatrémie je nadbytek hyponatremické tekutiny, tzn. nadbytek vody je větší než nadbytek soli. Výsledkem je diluční a také distribuční hyponatrémie (expanze nejen ECT, ale i třetího prostoru), stav zásob Na+ v organismu je zvýšený.
- Hypovolemická hyponatrémie s nízkou U-Na svědčí pro extrarenální příčinu ztrát tekutiny s vysokým obsahem Na+ (GIT, kůže).
- Hypovolemická hyponatrémie s vysokou U-Na je přítomna při renální příčině ztrát iontů sodíku provázených neproporčně menšími ztrátami vody; příčinou je porucha zpětné tubulární resorpce iontů sodíku v nefronech.
- Hypervolemická hyponatrémie s nízkou U-Na provází primárně non-renální edémové stavy (srdeční insuficienci, jaterní insuficienci) a část pacientů s nefrotickým syndromem, kdy je primárně snížený efektivní cirkulující objem, což vede k aktivaci natriumretenčních mechanismů a zvýšení celkového objemu tekutin a soli v organismu.
- Hypervolemická hyponatrémie s vysokou U-Na je odrazem renální retence iontů sodíku při akutním anebo chronickém selhání funkce ledvin, kdy v důsledku výrazného snížení GFR i přes maximální omezení zpětné resorpce iontů sodíku v tubulech dochází k pozitivní bilanci sodných iontů.
- Izoosmolální hyponatrémie vzniká při hyperlipidémii nebo hyperproteinémii – viz výše pseudohyponatrémie.
- Hyperosmolální hyponatrémie je výsledkem přítomnosti jiného efektivního faktoru než sodíku s následným vzestupem efektivní osmolarity, která vede k přesunu vody z buněk do ECT a z toho plynoucímu poklesu plazmatické koncentrace iontů sodíku. Typickým příkladem je diabetes mellitus, kdy se celková efektivní osmolarita zvyšuje vlivem hyperglykémie. Glukóza při diabetu je zvýšeně produkována játry, kde její syntéza není tlumena, a navíc nemůže vstupovat do buněk závislých na inzulínu, obojí proto, že je absolutní anebo relativní nedostatek tohoto hormonu. Vztah mezi vzestupem glykémie a poklesem natrémie není lineární, nicméně pro snadné zapamatování můžeme počítat s poklesem natrémie o 2 mmol/l na každých 5 mmol/l zvýšení glykémie.
- Z praktického terapeutického hlediska je ale přínosnější posouzení, o kolik se zvýší natrémie při léčbě hyperglykémie ? K odhadu slouží rovnice PvNa = PaNa/[1 – 0,002 x (PaG – PcG)], kde PvNa je předpokládaná výsledná natrémie po korekci hyperglykémie, PaNa je aktuální natrémie, PaG aktuální glykémie pacienta před zahájením léčby, PcG je cílová glykémie na konci léčby a 0,002 je koeficient vyjadřující k jaké objemové změně (v litrech) dojde v daném kompartmenu přesunem vody mezi ECT a ICT při změně koncentrace efektivního solutu (zde glukózy) o 1 mmol/l.
- Hypoosmolální hyponatrémie neboli „pravá“ hyponatrémie, které příčinou je nepoměr mezi zásobami Na+ v ECT a objemem ECT ve prospěch objemu.
Příčiny hypovolemické hyponatrémie
Extrarenální ztráty iontů sodíku
- Gastrointestinální ztráty. Trávicí šťávy mají různou koncentraci iontů sodíku, ale obvykle jsou hyponatremické, takže spíše vedou k hypernatremické dehydrataci než dehydrataci hyponatremické. Sklon k hyponatrémii mívají sekreční průjmy (např. cholera) s vysokým obsahem Na+ v průjmové tekutině. Častější příčinou hyponatrémie při průjmu, zvracení, píštělích anebo ileózních stavech je až nesprávně vedená rehydratační terapie, kdy jsou hrazeny pouze ztráty vody, kdežto ztráty iontů sodíku jsou hrazeny málo nebo vůbec (např. pitím černého čaje nebo roztokem 5% glukózy). Některé léky mohou narušit resorpci iontů sodíku v tlustém střevě - například iritačně působící laxativa (pikosulfát, antrachinonové glykosidy); šetrnější jsou osmotická laxativa (laktulóza, síran hořečnatý).
- Kůže. Stejně jako v případě GIT ztrát je i pot obvykle tekutinou s nižší koncentrací Na+, než má plazma. K hyponatrémii proto dochází jen při excesivním pocení u déle trvající horečky anebo při vytrvalostní sportovní aktivitě, kdy průběžná úhrada tekutin neobsahuje dostatek soli, a dále u neaklimatizovaných osob v horkých provozech anebo podnebných pásech. Rizikovou skupinou pro rozvoj hyponatrémie i při běžném pocení jsou děti a lidé s cystickou fibrózou [12], u kterých je obsah soli v potu vysoký následkem genetické poruchy.
Renální ztráty iontů sodíku
- Osmotická diuréza. Vzniká v situacích, kdy se do moči přes glomerulární filtr anebo vystupňovanou tubulární sekrecí dostává efektivní iont nebo solut, který se v průběhu tubulů nedostatečně anebo vůbec neresorbuje. Tento solut pak na sebe váže vodu, která s sebou strhává sodíkové ionty a ostatní ionty. Navíc zrychlený průtok moči distálním nefronem neumožňuje dostatečnou zpětnou resorpci iontů sodíku, i když dehydratace aktivuje systém renin-angiotenzin-aldosteron. Typickým příkladem situace provázené osmotickou diurézou je diabetická ketoacidóza, kdy oním efektivním solutem je jak glukóza, tak ketolátky. Léčebně se osmotické diurézy užívá u edémových stavů, kdy ji vyvoláváme manitolem.
- Diuretika, která přímo inhibují zpětnou resorpci soli v průběhu tubulů, logicky vedou k hyponatrémii. Kličková diuretika působí na úrovni tlusté vzestupné části Henleovy kličky, kde blokují Na+/K+/2Cl- kotransportér na luminální membráně. Co do diuretického účinku (množství moči za jednotku času) jsou sice kličková diuretika nejsilnější (20 % ultrafiltrátu se resorbuje v Henleově kličce), ale nejčastější příčinou hyponatrémie jsou thiazidová diuretika, která blokují Na+/Cl- kotransportér (NCC) ve stočené části distálních tubulů. Důvodem je fakt, že thiazidová diuretika nenarušují koncentrační schopnost ledvin, takže diuréza je po nich sice menší, ale s relativně větší natriurézou. Naproti tomu porucha resorpce soli v Henleově kličce po podání kličkových diuretik vede ke snížení hypertonicity dřeně ledvin, a tím větší, ale proporcionálnější diuréze (měřeno poměrem ztrát soli a vody). Pro kličková a thiazidová diuretika současně platí, že vedou jak k hyponatrémii, tak k hypokalémii. Tím se liší od tzv. kalium-šetřících diuretik, která působí až v druhé části distálních tubulů a vedou ke kombinaci hyponatrémie a hyperkalémie. Mechanismus jejich účinku je dvojí – buď kompetují s aldosteronem v jeho vazbě na receptory v tubulárních buňkách (spironolakton), nebo ruší účinek aldosteronu blokádou ENaC kanálů v distálním nefronu s následným snížením reabsorpce iontů sodíku, a tím i exkrece iontů draslíku (amilorid, triamteren).
- Salt-losing nefritis. Tento pojem je spíše syndromologický a zahrnuje všechny nefropatie charakterizované vysokými ztrátami iontů sodíku močí. Může se jednat o polyurickou formu akutního renálního selhání (ARS), nebo o polyurickou fázi oligoanurického ARS (viz např. kapitola Patofyziologie a klinické aspekty akutního poškození a selhání ledvin [13]), následek chronické tubulointersticiální nefritidy, postobstrukční polyurii, st. p. transplantaci ledvin apod.
- Deficit mineralokortikoidů. Chybějící sekrece nebo snížený účinek aldosteronu může mít řadu příčin. Výsledkem je zvýšený odpad sodíku močí, hyponatrémie, hyperkalémie se sklonem k metabolické acidóze a dehydratace s rizikem rozvoje šoku a smrti organismu. Primární hypokorticismus vzniká při postižení kůry nadledvin, jako je tomu u Addisonovy choroby, což je autoimunitní postižení, které vede k chronickému hypokorticismu se sníženou sekrecí jak aldosteronu, tak kortisolu a nadledvinových androgenů. Akutní adrenokortikální insuficience může vzniknout při dekompenzaci u pacienta trpícího Addisonovou nemocí, který se dostane do stresové situace (nemoc, operace atp.), nebo při akutním krvácení do nadledvin při krvácivých chorobách, při traumatu (novorozenci) anebo jako Waterhouse-Friderichsenův syndrom v rámci meningokokové sepse. Sekundární hypokorticismus při nedostečné sekreci ACTH adenohypofýzou k hypoaldosteronismu nevede, protože přímá sekrece aldosteronu na rozdíl od sekrece kortisolu je na stimulaci ACTH závislá jen minimálně. Některé z forem kongenitální adrenální hyperplázie (CAH) mohou být provázeny solnou poruchou z nedostatku aldosteronu. Nejčastěji jde o deficit 21-hydroxylázy, resp. její SW (salt wasting formu), kdy je nedostatečná syntéza jak gluko-, tak mineralokortikoidů, a naopak je přebytek nadledvinových androgenů, které u děvčátek vedou k virilizaci genitálu a u chlapců k předčasné pubertě. Pseudohypoaldosteronismus je genetická porucha účinku aldosteronu. Pacienti nemají nízkou, ale naopak vysokou hladinu aldosteronu, ale příznaky jsou stejné jako u hypoaldosteronismu. U typu I autosomálně recesivně dědičného pseudohypoaldosteronismu jde o inaktivační mutace některé z podjednotek epiteliálního sodíkového kanálu (ENaC) zajišťujícího resorpci Na+ vyvolanou aldosteronem v distálním nefronu. U typu II autosomálně dominantně dědičného pseudohypoaldosteronismu jde o mutaci mineralokortikoidního receptoru nacházejícího se v jádře buněk distálních tubulů a sběracích kanálků.
- Cerebral salt wasting syndrom (CSWS) může vzniknout při poškození mozku různé etiologie - neurochirurgickým zákrokem, kraniotraumatem, mozkovým nádorem, cévní mozkovou příhodou atp. Patogeneze tohoto syndromu není zcela jasná. Může se uplatňovat vystupňovaná sekrece natriuretických faktorů (ANF, BNF), tlaková diuréza , anebo změna renálního průtoku krve vlivem změny tonu sympatiku. Následky jsou ale stejné - vystupňování natriurézy, polyurie, hyponatrémie, dehydratace. Sekundárně k efektivní hypovolémii je zvýšená sekrece ADH (přes baroreceptorové stimuly). Hyponatrémie se zvýšenou sekrecí ADH je rovněž známkou SIADH, který se mimo jiné může rozvíjet v souvislosti s etiologicky pestrými poškozeními mozku, ale u SIADH je nepřiměřeně vysoká hladina ADH primární příčinou, kdežto při CSWS je až následkem. Oba syndromy je nutné od sebe rozlišit, protože léčba je zcela odlišná (diferenciální diagnostiku probereme u SIADH).
- Nediuretická farmaka. Celá řada léků, ač nejsou primárně diuretiky, může poškozovat schopnost ledvin zpětně resorbovat ionty sodíku, a tak vést hyponatrémii. Aminoglykosidová antibiotika působí nefrotoxicky, protože při vylučování do moči jsou vychytávána jako anionty přes transportéry pro bázické oligopeptidy v buňkách proximálních tubulů. Tyto buňky je ale nejsou schopny metabolizovat, což vede ke kumulaci aminoglykosidů v proximálních tubulech s jejich následným poškozením. Rovněž tak tubulotoxicky působí například cisplatina anebo cyklosporin A. Hyponatrémie může vzniknout i účinkem léků zasahujících do regulace Na+ homeostázy, například léky interferující se systémem RAS, jako jsou ACE inhibitory nebo blokátory AT receptorů. Ke zvýšeným ztrátám iontů sodíku močí vedou i beta-blokátory, a to snížením sekrece reninu v juxtaglomerulárním aparátu. Nesteroidní antiflogistika snižují sekreci reninu, ale také aldosteronu blokádou syntézy prostaglandinů (PG); jejich hyponatremizující účinek se ovšem projeví jen tam, kde je syntéza PG vystupňována, tak jako tomu je u hypertoniků. Řada léků může vést ke vzniku SIADH, ale tento syndrom patří mezi euvolemické hyponatrémie, a proto ho zmíníme až v další části.
Příčiny euvolemické hyponatrémie
Syndrom nepřiměřené sekrece antidiuretického hormonu (SIADH)
Syndrom nepřiměřené sekrece antidiuretického hormonu (SIADH) představuje klinicky nejčastější a nejvýznamnější příčinu euvolemické hyponatrémie (jeho výskyt je udáván u asi 30 % hospitalizovaných pacientů).
Patogeneze: Pojem „euvolemický“ je v tomto případě poněkud zavádějící, protože patogenetickým podkladem stavu je nepřiměřeně vysoká sekrece ADH, která neodpovídá akuální osmolalitě panující v ECT. Hypotalamický osmostat tedy má snížený práh pro vylučování ADH, a osmolalita tělních tekutin je udržována na nové, nižší úrovni. Výsledkem je retence bezsolutové neboli „čisté“ vody s následným rozvojem diluční hyponatrémie. Po klinické stránce ale pacient na první pohled nemá známky hyperhydratace. Chybí edémy, protože hyponatrémie vede k přesunu vody do ICT. Současně mírná volumoexpanze sekundárně aktivuje natriuretické mechanismy. Ztráty iontů sodíku i vody ledvinami se tím zvyšují, což vede k úplné anebo téměř úplné normalizaci objemu ECT (tedy „euvolémii“).
Etiologie: SIADH je syndromem s pestrou škálou příčin, které si můžeme rozdělit do několika podskupin:
- Poškození CNS traumatem, zánětem, neúrazovým krvácením, mozkovým nádorem, neurochirurgickým zákrokem.
- Zhoubné nádory, a to i lokalizované mimo CNS. Typickým příkladem je malobuněčný karcinom plic s ektopickou sekrecí ADH.
- Příčiny hrudní, jako jsou bronchopneumonie, astmatický záchvat, atelektáza, pneumothorax, akutní respirační selhání anebo umělá plicní ventilace; mechanismus vzniku SIADH je zde neznámý.
- Trauma, chirurgický zákrok – zejména břišní a hrudní operace. Zde by mohla být patogenetickým pojítkem bolest a po jejím přepojení přes limbický systém zvýšení sekrece ADH.
- Léky podobné ADH, jako je desmopresin nebo oxytocin, které stimulují V2 receptory v ledvinách.
- K SIADH ovšem může vést terapie celou řadou jiných léků, které nějakým mechanismem zvyšují uvolňování či účinek ADH. Zejména se jedná o léky ze skupiny antiepileptik (valproát, carbamazepin), imunosupresiv (cyklofosfamid, methotrexát), cytostatik (cisplatina, vinkristin, vinblastin), psychofarmak (inhibitory MAO, SSRI, haloperidol), antiarytmik (amiodaron), analgetik (opiáty, NSA), dále interferon α, interferon γ a mnohé další.
- Vrozený SIADH, a to buď „hypotalamický“ při mutaci genu TRPV4, který vede k poruše vnímání osmolality centrálními osmoreceptory, nebo „nefrogenní“ kdy jde o aktivační mutaci V2 receptoru v ledvinách (hladina ADH zde bude snížená, na rozdíl od ostatních forem SIADH).
- Idiopatický SIADH, kdy je příčina nejasná.
Diagnostická kritéria SIADH zůstávají v platnosti tak jak je v r. 1967 stanovili Bartter se Schwartzem:
 [14]
[14]Diagnostická kriteria SIADH
- Hypotonická hyponatrémie (S-Na < 130 mmol/l, S-osm < 275 mmol/kg) tedy „pravá“ hyponatremie
- Nepřiměřeně vysoká osmolalita moči (U-osm > 100 mmol/kg). Měřeno absolutními hodnotami je U-osm nízká, ale vzhledem k osmolalitě plazmy je neadekvátně vysoká, protože při S-osm pod 275 mmol/kg by moč měla být maximálně „naředěná“ s U-osm blížící se hodnotě 50 mmol/kg.
- Klinická euvolémie, tzn. pacient nemá ani známky dehydratace, ani nejsou přítomny edémy.
- Zvýšená koncentrace iontů sodíku v moči (U-Na nad 20 mmol/l) je důležitým diferenciálně diagnostickým kritériem sloužícím k odlišení stavů se sníženým efektivním cirkulujícím objemem, kde naopak je vlivem aktivace systému RAA U-Na snížená.
- Vyloučení jiných příčin hyponatrémie. SIADH je diagnózou per exclusionem. Je zejména nutné vyloučit vliv diuretické terapie, poruchy funkce štítné žlázy, nadledvin, psychogenní polydipsii. Laboratorně vypadá podobně jako SIADH syndrom CSWS. Klinický rozdíl u pacientů s CSWS může být diskrétní, zejména pokud je pacient léčen infúzemi s obsahem iontů natria. Významným laboratorním kritériem je v takovém případě exkreční frakce vody (FEH2O) a natria (FENa), které jsou u SIADH snížené až normální, kdežto u CSWS jsou vždy zvýšené.
Principy léčby SIADH:
- Restrikce tekutin, tzn. pacient přijímá méně tekutin než vymočí plus započtení perspiratio insensibilis.
- Hypertonický roztok NaCl se používá jen v akutní fázi terapie život ohrožující hyponatrémie. Následně se přechází na zvýšený perorální přísun soli. Dochází ke zvýšení natrémie, přebytek vody se přesunuje z buněk do ECT.
- To v kombinaci s podáním kličkových diuretik, jako je furosemid, vede ke zvýšení diurézy.
- Novou možností léčby je použití antagonistů vazopresinových receptorů. Zejména se používá neselektivní conivaptan (blokuje jak V2 receptory v ledvinách, tak V1 receptory v cévách). Limitem teoreticky výhodnějších selektivních V2 blokákorů (tolvaptan, satavaptan, lixivaptan) je kromě vyšší ceny zvýšený pocit žízně a nebezpečí příliš rychlého vzestupu natrémie.
- Mezi alternativní léčebné přístupy patří podání urey, která zvýší vylučování bezsolutové vody ledvinami, dále derivát antibiotika tetracyklinu demeklocyklin, který svým nefrotoxickým účinkem indukuje stav podobný diabetes insipidus (proto je nutné během léčby monitorovat renální funkce).
- Odstranění příčiny SIADH, pokud je to možné, představuje kauzální léčbu.
- Nevhodným terapeutickým přístupem u SIADH je podávání izonatremických infúzí, protože zatímco sodík se vyloučí, voda je zadržena a hyponatremie se zhorší !
Primární polydipsie
Hypotyreóza
Izolovaný deficit glukokortikoidů
Nízký přísun soli
Hyponatrémie „maratonských běžců“
Příčiny hypervolemické hyponatrémie
Selhání ledvin
Srdeční selhání
Jaterní selhání
Příznaky hyponatrémie
Principy terapie hypotonické hyponatrémie
 [18]
[18]- Zásady rychlosti korekce hyponatremií

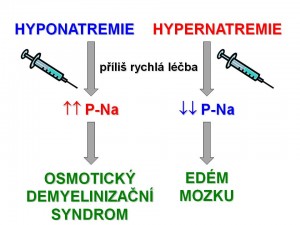 [19]
[19]- Rizika terapie hyponatremií a hypernatremií
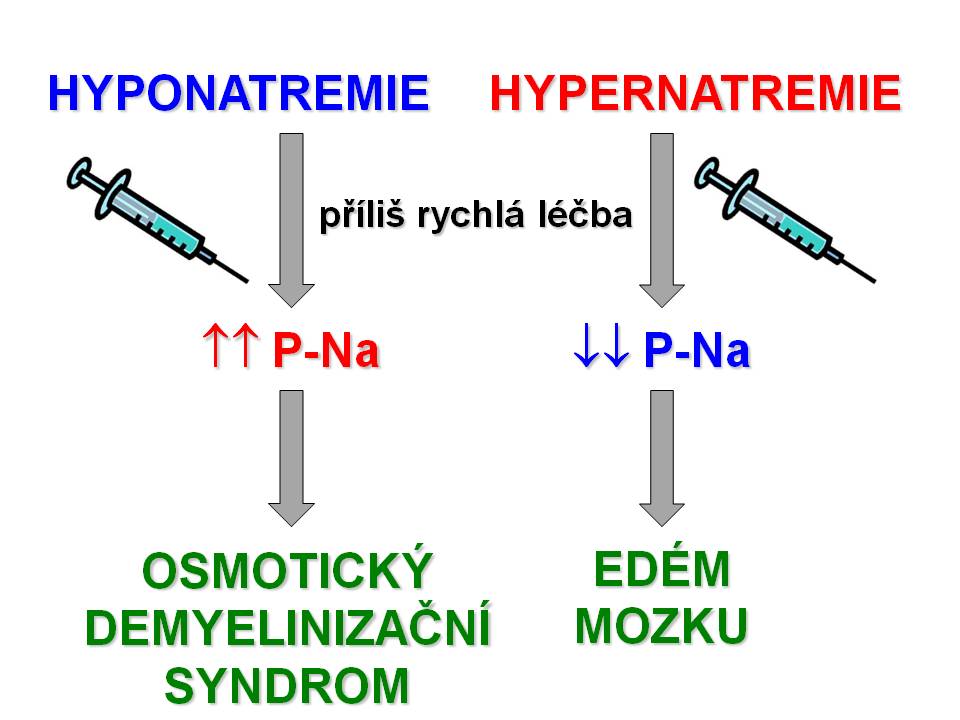
Definice hypernatrémie
Výskyt hypernatrémie
Klasifikace hypernatremických stavů
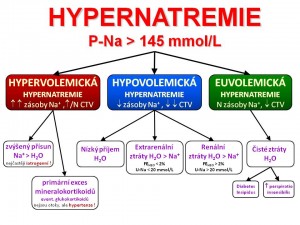 [20]
[20]- Příčiny hypernatremie
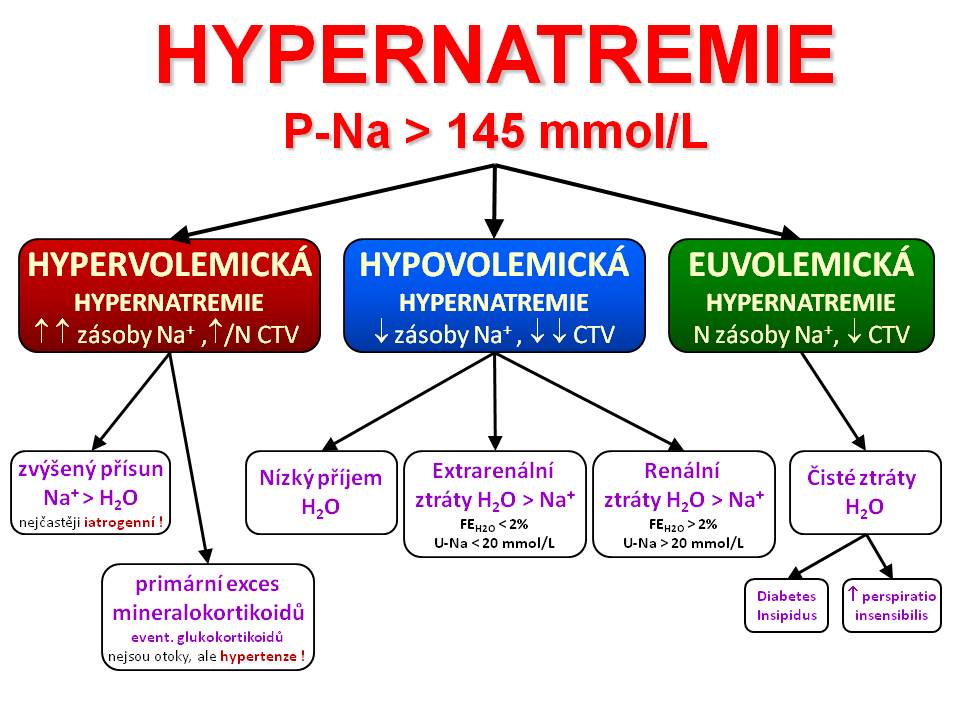
Hypovolemická hypernatrémie
Zvýšená plazmatická koncentrace Na+ se kombinuje se sníženým objemem ECT. Vyšetřením pacienta nacházíme klinické a laboratorní známky dehydratace. Obecnou příčinou tohoto typu hypernatrémie je ztráta hyponatremické tekutiny. Vzniká tím, že ztráty vody jsou větší než ztráty Na+; celkové zásoby Na+ v organismu jsou snížené.
Izovolemická hypernatrémie
Zvýšená plazmatická koncentrace Na+ se kombinuje s téměř normálním objemem ECT. Vyšetřením pacienta nenacházíme klinicky zjevné známky hyperhydratace ani dehydratace. Obecnou příčinou tohoto typu hypernatrémie je buď nedostatečný příjem vody anebo zvýšené ztráty čisté vody, přičemž celotělové zásoby Na+ jsou normální. Pokud má pacient zachovaný přístup k tekutinám, pak vzniklá hypernatrémie stimuluje žízeň, což vede k náhradě objemu, a pacient pak zůstává izovolemický.
Hypervolemická hypernatrémie
Zvýšená plazmatická koncentrace Na+ se kombinuje se zvýšeným objemem ECT. Vyšetřením pacienta nacházíme klinické a laboratorní známky hyperhydratace. Obecnou příčinou tohoto typu hypernatrémie je nadbytek hypernatremické tekutiny. Nadbytek iontů sodíku je za těchto okolností větší než nadbytek vody jako následek zvýšeného přívodu nebo sníženého výdeje soli z organismu. Celkové zásoby Na+ v těle jsou každopádně zvýšené.
Příčiny hypovolemické hypernatrémie
Gastrointestinální trakt
Může být jednou z extrarenálních cest ztrát. V případě lehčích infekčních průjmů a zvracení se složením jedná o tekutiny hypotonické s nízkým obsahem iontů natria, takže výsledkem je hypernatrémie. Stolice bývá hypotonická rovněž u osmotických průjmů. Příkladem je vrozený nebo získaný (častěji) deficit laktázy, kdy pro nedostatek enzymu laktázy v kartáčovém lemu enterocytů pacient není schopen v tenkém střevě štěpit mléčný disacharid laktózu. Nerozštěná laktóza není ze střeva vstřebatelná, osmoticky natahuje do lumina střeva vodu, což zvyšuje motilitu; laktóza pak postupuje do tlustého střeva, kde probíhá její fermentace střevní mikroflórou za vedlejší produkce kvasných plynů a mléčné kyseliny. Výsledkem je meteorismus se vzedmutím břicha, průjem, stolice jsou napěněné, vodnaté, kysele páchnoucí. Na osmotickém principu fungují i některá projímadla používaná v léčbě zácpy, jako jsou třeba lactulosa a solná projímadla. Stejně tak může vést k průjmu přijetí velkého množství ovoce anebo džusů, které obsahují fruktósu (ta se sice v tenkém střevě vstřebává, ale při přijetí velkého množství je kapacita transportního systému přetížena). K úniku hypotonické střevní tekutiny také může docházet přes různé píštěle anebo přes stomii. Vzácnější příčinou hypernatremické dehydratace může být peritoneální dialýza.
Kůže
Pocení v horkém prostředí nebo při horečce vede ke ztrátám hypotonické tekutiny kůží, a pokud není hrazeno zvýšeným příjmem tekutin, pak je výsledkem hypernatremická dehydratace.
Ledviny
Ke ztrátě hypotonické tekutiny renální cestou dochází při osmotické diuréze (glukóza, ketolátky, manitol atp.), při použití diuretik (zejména kličkových), u některých renálních postižení (polyurická fáze při CHRI, stavy po odstranění obstrukce).
Absolutně nedostatečný příjem vody
Hrozí u pacientů, kteří mají z nějakého důvodu znemožněn přístup ke zdroji vody, tedy malé děti, staří, bezmocní anebo s poruchou vědomí.
Příčiny izovolemické hypernatrémie
Relativně nedostatečný příjem vody
Diabetes insipidus (DI)
Patogeneticky se rozlišují dva typy:
Centrální diabetes inspidus (CDI) při nedostatečné tvorbě ADH v hypotalamu. Etiologie: Příčinou je obvykle získané poškození mozku resp. oblasti hypotalamu a neurohypofýzy, jako jsou trauma, krvácení, hypoxie-ischémie, infekce, neurochirurgický zákrok, nádor (kraniofaryngeom, nádor vycházející z hypofýzy, leukémie/lymfom, metastáza jiného nádoru) , granulomatózy (sarkoidóza, histiocytóza), radiační záření. Vzácně jde o vrozenou příčinu, jako je septo-optická dysplázie, Wolframův syndrom nebo mutace genu pro ADH-neurofyzin. U části pacientů příčinu neodhalíme a pak hovoříme o idiopatickém CDI; někdy lze předpokládat autoimunní etiologii.
Nefrogenní diabetes insipidus (NDI) při nedostatečné účinnost ADH v ledvinách. Etiologie: Mezi získané příčiny patří: Renální choroby (CHRI, polycystické ledviny, obstrukční uropatie).Někdy je NDI indukován léky, jako např. demeklocyklin (viz SIADH), soli Li+, amphotericin B, kolchicin , ale také kličková diuretika (resorpce soli v Henleově kličce je jednou ze základních podmínek pro udržení osmotického koncentračního gradientu). Patogeneticky zajímavou příčinou NDI je deficit draslíku a hypokalémie, kde jsou možná dvě vysvětlení - deficit K+ vede (a) k narušení tvorby a udržení hyperosmolarity dřeně ledvin, nebo (b) k „rezistenci“ na účinek ADH ve sběracích kanálcích. Další iontovou odchylkou vedoucí k polyurii je hyperkalciurie. Její účinek spočívá ve změněné funkci CaSR (calcium sensing receptor) na luminální membráně tubulárních buněk, na který se vážou ionty vápníku. To vede ke snížení hladiny cAMP, který je 2. poslem účinku ADH. Vrozené formy NDI jsou raritní. Mezi nimi 90 % tvoří mutace V2 receptoru pro ADH v tubulárních buňkách sběracích kanálků. Dědičnost je gonosomálně recesivní. Zbývajících 10 % vrozených NDI připadá na autozomálně recesivní mutaci genu pro aquaporin-2, tj. protein vodního kanálku na apikální membráně tubulárních buněk sběracích kanálků.
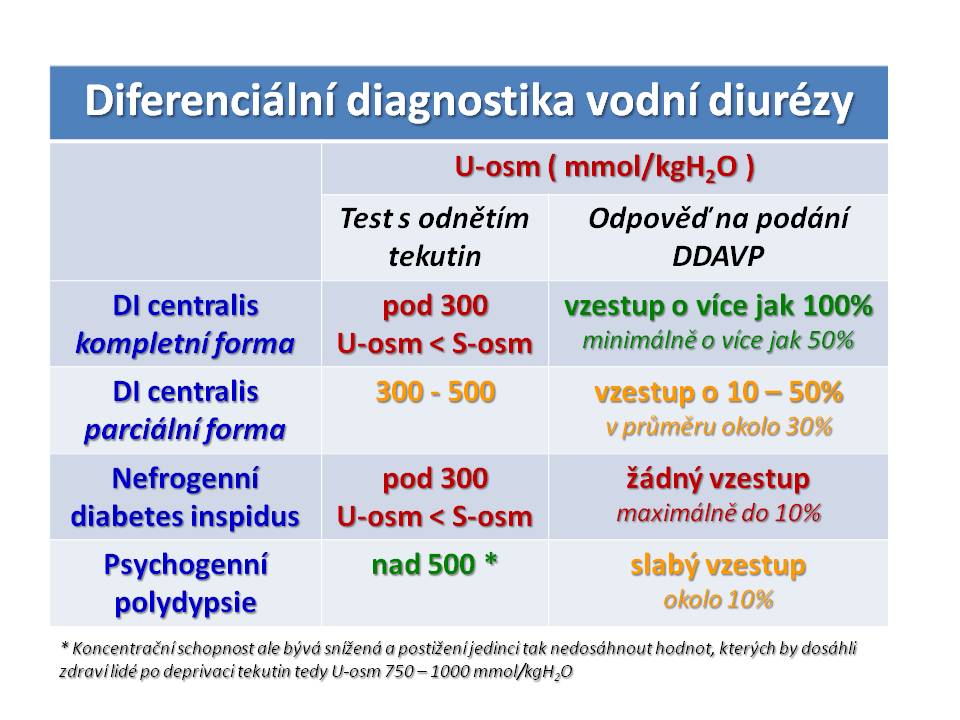 [21]
[21]Diferenciální diagnostika polyurie s nízkou osmolalitou moči
Zvýšené ztráty čisté vody
Příčiny hypervolemické hypernatrémie
Hyperosmolární hypernatremické infúze
K léčbě metabolických acidóz je mnohdy zbytečně (například v léčbě diabetické ketoacidózy) využíván hyperosmolární 4,2% nebo dokonce 8,4% roztok NaHCO3. Rovněž nevhodné množství 3% NaCl použitého v léčbě hyponatrémie může vést k přestřelení do hypernatrémie. Hypertonické salinické roztoky se také užívají v léčbě intrakraniální hypertenze po úrazech mozku, a proto i zde může nastat hypernatrémie.
Enterální nadměrný přívod sodíku s hypernatrémií
Je popisován při enterální výživě hyperosmolárními roztoky, při vysokoproteinové dietě, při laváži žaludku hypertonickým roztokem NaCl nebo použití roztoku soli jako emetika při intoxikacích.
Pití mořské vody
Trosečnící na moři donucení pít slanou vodu jsou ohroženi výraznou hyperosmolaritou z hypernatrémie.
Primární hyperaldosteronismus neboli Connův syndrom
Jeho příčinou je hyperplázie nebo hormonálně aktivní adenom, vzácně karcinom kůry nadledvin. Nadprodukce mineralokortikoidu vede ke zvýšenému zadržování iontů sodíku současně se zvýšeným vylučováním iontů draslíku. Zatímco ale hypokalémie společně s metabolickou alkalózou jsou u této diagnózy typickým laboratorním nálezem, hypernatrémie bývá vzácná. Retence iontů sodíku ledvinami totiž vede i k zadržování vody, a tím se hypernatrémie mírní. Každopádně dochází k expanzi volumu, a roste krevní tlak, který a mechanismem tlakové natriurézy postupně zvyšuje výdej sodíku. Mluvíme o tzv. fenoménu úniku, kdy v ledvinách ionty sodíku postupně s rostoucím krevním tlakem unikají zpod reabsorpčního vlivu aldosteronu; krevní tlak narůstá tak dlouho, dokud nedojde k obnově vyrovnané bilance příjmu a výdeje iontů sodíku, a tím i tekutin. „Obětí“ se stává krevní tlak, protože arteriální hypertenze je z pohledu přežití organismu podstatně menší problém než nerovnováha v příjmu soli a vody. Pomocným diagnostickým kritériem je poměr koncentrace iontů sodíku a draslíku v moči, U-Na/U-K, který je v tomto případě menší než 1,0 (normálně bývá okolo 2:1).
Cushingův syndrom
Příčinou je zde (a) nadbytek endogenního kortizolu (centrální typ s nadprodukcí ACTH adenohypofýzou, periferní typ s hyperplázií nebo nádorem kůry nadledvin, popř. paraneoplastický typ s ektopickou tvorbou ACTH), nebo (b) nadbytek exogenních kortikoidů,
Kortisol je sice jsou slabý mineralokortikoid, ale při vysokých hladinách se nestačí v ledvinách odbourávat účinkem 11-beta-HSD2 a váže se na mineralokortikoidní receptory v tubulárních buňkách a výsledkem je – obdobně jako u primárního hyperaldosteronismu – volumová hypertenze a zvýšené zásoby Na+ v těle, méně často hypernatrémie.
Příznaky hypernatrémie
Stejně jako u hyponatrémie závisí i v případě hypernatrémie projevy této poruchy na rychlosti vzniku a na stupni hypernatrémie. Stejně jako u hyponatrémie jsou i příznaky hypernatrémie dány postižením mozku.
Základním patogenetickým mechanismem v rozvoji příznaků akutní hypernatrémie je exsikace mozkových buněk, která vzniká přesunem vody z buněk do hypertonického extracelulárního prostředí. Jelikož hypernatrémie znamená hyperosmolaritu, je hlavním příznakem žízeň. Ovšem nepřítomnost žízně při hypernatrémii signalizuje poruchu centra žízně v hypotalamu, a tím i vysoké riziko rychlého rozvratu vnitřního prostředí. Jinak mohou být iniciální příznaky hypernatrémie nenápadné a nespecifické - nechutenství, letargie, nauzea a svalová slabost nebo neklid, podrážděnost, zmatenost apod. Objektivně lze nalézt hyperreflexii. Tyto příznaky pak při těžší hypernatrémii mohou přejít v křeče a v kóma. Situaci může komplikovat subarachnoidální nebo subkortikální krvácení, které vzniká následkem dehydratace mozku provázené vzájemnou separací mozkových obalů od sebe s průvodní rupturou přemosťujících mozkových žil, nebo venózní trombóza.
Při chronické hyponatrémii mají mozkové buňky dostatek času se adaptovat tím, že zvyšují množství intracelulárních iontů a solutů. Díky tomu jsou omezeny ztráty vody z buněk a objemu buněk, a tím i symptomatologie hypernatrémie je chudá.
Principy terapie hypernatremických stavů
Předpokladem úspěšné léčby hypernatrémie je odhalení její příčiny a mechanismu vzniku. Správné zhodnocení klinických a laboratorních příznaků v kombinaci s rozpoznáním příčiny umožňuje také zhodnotit stav zásob Na+ v organismu, protože hypernatrémie může provázet i situace se sníženým celkovým množstvím zásob celotělového Na+.
- [19]
- Rizika terapie hyponatremií a hypernatremií
Vlastní korekce hypernatrémie pak je závislá na rychlosti rozvoje poruchy. Při akutní hypernatrémii by korekce měla proběhnout v časovém horizontu 24 hodin. Rychlost poklesu natrémie může být 1 mmol/l za hodinu.
Naopak při chronické hypernatrémii je zapotřebí počítat s adaptací mozkových buněk. Korekce by měla probíhat pomalu, a je-li to potřebné, i déle než 48 hodin. Doporučená rychlost poklesu při chronické hypernatrémii nebo u hypernatrémie, jejíž délka trvání je nejasná, by neměla přesáhnout 10 – 12 mmol/l za 24 hodin, tedy maximálně 0,5 mmol/l za hodinu. V terapii se používají kombinace roztoků 5% glukózy a iontových roztoků s koncentrací Na+ nižší, než je aktuální hyperatrémie (1/2, 2/3, 1/1 F, přičemž 1/1 F má koncentraci Na+ 155 mmol/l). Při hypovolemické hypernatrémii stačí doplnit chybějící objem pomocí těchto roztoků.
Co se týče množství čisté vody potřebné k uhrazení jejího deficitu, lze použít následující vzorec: Deficit čisté H2O = [(P-Navstupní /140) – 1] x objem celkové tělesné vody = [(P-Navstupní /140) - 1] x hmotnost (kg) x 0,6 (u žen 0,55). Ovšem tento vzorec počítá s „normálním“ objemem CTV, takže je třeba vzít v úvahu, že v případě hypovolémie je deficit vody větší než vypočítaný. Léčba euvolemické hypernatrémie při diabetes inspidus byla popsána výše. U hypervolemické hypernatrémie kombinujeme výše uvedenou infuzní terapii s podáním diuretik k vyloučení přebytečného objemu soli a vody.
Použitá literatura a literatura k dalšímu studiu
- BAGSHAW SM, TOWNSEND DR, McDERMID RC. Disorders of sodium and water balance in hospitalized patiens. Can J Anesth. 2009, 56: 151-167
- BERNE RB, LEVY MN, KOEPPEN BM, STANTON BA. Physiology, 5th edition, Mosby 2004, pp. 643-683
- GUYTON AC, HALL JE. Textbook of medical physiology, 11th edition, Elsevier Saunders 2006, pp. 327-364
- HULÍN I. et al., Patofyziológia, 7.vydání. Bratislava SAP 2009, str. 256-262
- JABOR a kol. Vnitřní prostředí. 1.vydání, Grada 2008, str. 21–45
- LEWIS JL. Hypernatremia. www.merckmanuals.com
- Makaryus AN, McFarlane SI. Diabetes insipidus: diagnosis and treatment of a complex disease. Cleve Clin J Med. 2006 Jan;73(1):65-71.
- MASOPUST J. Klinická biochemie, požadování a hodnocení biochemických vyšetření, I,II část. Karolinum, Praha 1998, str. 218-227, 731-751
- McCANCE KL, HUETHER SE, BRASHERS VL, ROTE NS. Pathophysiology: the biological basis for disease in adults and children, 6th edition, Mosby Elsevier 2010, pp. 96-106
- Pillai BP, Unnikrishnan AG, Pavithran PV. Syndrome of inappropriate antidiuretic hormone secretion: Revisiting a classical endocrine disorder. Indian J Endocrinol Metab. 2011 Sep;15 Suppl 3:S208-15.
- ROSE BD. Renal actions of dopamine. www.uptodate.com
- SILBERNAGL S, DESPOPOULOS A. Atlas fyziologie člověka. Grada Avicenum, Praha 1993, str. 132-143
- SCHÜCK O. Poruchy metabolismu vody a elektrolytů v klinické praxi. Grada, Praha 2000, str. 63-82
- ŠÍMA M. Poléková hyponatremie. Edukafarm FarmiNews. 2011, 4(2): 52-53
- Thomson SC, Blantz RC. Glomerulotubular balance, tubuloglomerular feedback, and salt homeostasis. J Am Soc Nephrol. 2008 Dec;19(12):2272-5
- VERBALIS JG. Brain volume regulativ in response to changes in osmolarity. Neuroscience. 2010 Jul 28;168(4):862-70
- VERBALIS JG, Disorders of body water homeostasis, Best Pract Res Clin Endocrinol Metab. 2003 Dec;17(4):471-503
- Verbalis JG, Goldsmith SR, Greenberg A, Schrier RW, Sterns RH. Hyponatremia treatment guidelines 2007: Expert panel recommendations. Am J Med. 2007 Nov;120(11 Suppl 1):S1-21
- VESELÝ J. Tlaková diuréza a arteriální hypertenze. Epava 2002
Article printed from Tvorba a ověření e-learningového prostředí pro integraci výuky preklinických a klinických předmětů na LF a FZV UP Olomouc: http://pfyziolklin.upol.cz
URL to article: http://pfyziolklin.upol.cz/?p=3695
URLs in this post:
[1] Image: http://pfyziolklin.upol.cz/wp-content/uploads/2012/03/Na-distribuce.jpg
[2] Image: http://pfyziolklin.upol.cz/wp-content/uploads/2012/03/Na-bilance.jpg
[3] Image: http://pfyziolklin.upol.cz/wp-content/uploads/2012/03/Na-PT.jpg
[4] Image: http://pfyziolklin.upol.cz/wp-content/uploads/2012/03/Na-HK.jpg
[5] Image: http://pfyziolklin.upol.cz/wp-content/uploads/2012/03/Na-DT-a-CD.jpg
[6] Image: http://pfyziolklin.upol.cz/wp-content/uploads/2013/06/FTV-RAA.jpg
[7] Image: http://pfyziolklin.upol.cz/wp-content/uploads/2012/03/Na-aldosteron-K.jpg
[8] Image: http://pfyziolklin.upol.cz/wp-content/uploads/2012/03/Na-tlakova-natriuresa.jpg
[9] Image: http://pfyziolklin.upol.cz/wp-content/uploads/2012/04/FTV-TK-diuresa.jpg
[10] Image: http://pfyziolklin.upol.cz/wp-content/uploads/2012/03/Na-AP-neuronu.jpg
[11] Image: http://pfyziolklin.upol.cz/wp-content/uploads/2012/03/Na-hypoNa-dfdg.jpg
[12] cystickou fibrózou: http://pfyziolklin.upol.cz/?p=1235
[13] Patofyziologie a klinické aspekty akutního poškození a selhání ledvin: http://pfyziolklin.upol.cz/?p=1285
[14] Image: http://pfyziolklin.upol.cz/wp-content/uploads/2012/03/SIADH-kriteria.jpg
[15] Laboratorní vyšetření ledvinových funkcí: http://pfyziolklin.upol.cz/?p=1550
[16] chronického renálního selhání: http://pfyziolklin.upol.cz/?p=1329
[17] srdečním selháním : http://pfyziolklin.upol.cz/?p=2293
[18] Image: http://pfyziolklin.upol.cz/wp-content/uploads/2012/03/Na-lecba-hypoNa.jpg
[19] Image: http://pfyziolklin.upol.cz/wp-content/uploads/2012/03/Na-rizika-lecby.jpg
[20] Image: http://pfyziolklin.upol.cz/wp-content/uploads/2012/03/Na-hyperNa-dfdg.jpg
[21] Image: http://pfyziolklin.upol.cz/wp-content/uploads/2012/03/DI-df-dg.jpg
Click here to print.