

Srdeční výdej
Srdeční výdej (minutový srdeční objem, cardiac output, CO) je součinem systolického neboli tepového objemu (stroke volume, SV) a srdeční frekvence (F) za minutu:
CO (ml/min) = SV x F
V levé komoře je na konci diastoly asi 110 – 120 ml krve (EDV; end-diastolický volum). Toto množství se při systole zmenší asi na 50 ml (ESV; end-systolický volum), takže asi 70 ml krve se při systole vypudí do aorty (SV nebo česky též SO; systolický objem). Normální ejekční frakce komory EF = SO/EDV tedy je asi 0,6 – 0,7. Při normální srdeční frekvenci F je normální srdeční výdej asi 5000 – 5600 ml/min, kolísá však v širokých mezích (3,7 – 6 l/min).
Srdce může jednorázově zvětšit systolický objem tím, že na úkor end-systolického komorového volumu zvýší ejekční frakci. End-systolický objem pak může poklesnout až na 10 – 20 ml. Aby však i při následujících stazích mohl systolický objem zůstat zvýšený, musí se srdce znovu adekvátně naplnit, tj. musí při diastole přijmout větší množství krve. Za to odpovídá žilní návrat, tedy – z daleko největší části – periferie, nikoliv srdce samo. Zdravé srdce může pojmout 150 – 180 ml end-diastolického volumu. Systolický objem se tak v případě potřeby může přibližně zdvojnásobit. Spolu s frekvencí, která se může efektivně zvýšit téměř třikrát, tak může srdce v rámci svých fyziologických rezerv zajistit až šestinásobný nárůst srdečního výdeje (tj. při zátěži může stoupnout na 25-30 litrů/min, u trénovaných jedinců ještě více).
Srdeční výdej coby hemodynamická veličina je při zachovaných funkčních rezervách srdce přímo určen jinou hemodynamickou veličinou – žilním návratem. Srdeční výdej SV, a s ním i krevní tok J, proto nejsou fyziologické primární, nezávislé, nýbrž sekundární, závisle proměnné hemodynamické veličiny. Platí to navzdory často mechanicky tradovanému pojetí srdce jako všemocného dominantního hráče v krevním oběhu.
Srdeční výdej určují čtyři následující faktory (obr. 1):
- Náplň srdce za diastoly - předtížení (preload) srdečního svalu;
- Odpor, s kterým se setkává krev vypuzovaná při systole komor do aorty - dotížení (afterload) srdečního svalu;
- Síla a rychlost stahu (kontraktilita) myokardu;
- Srdeční frekvence.
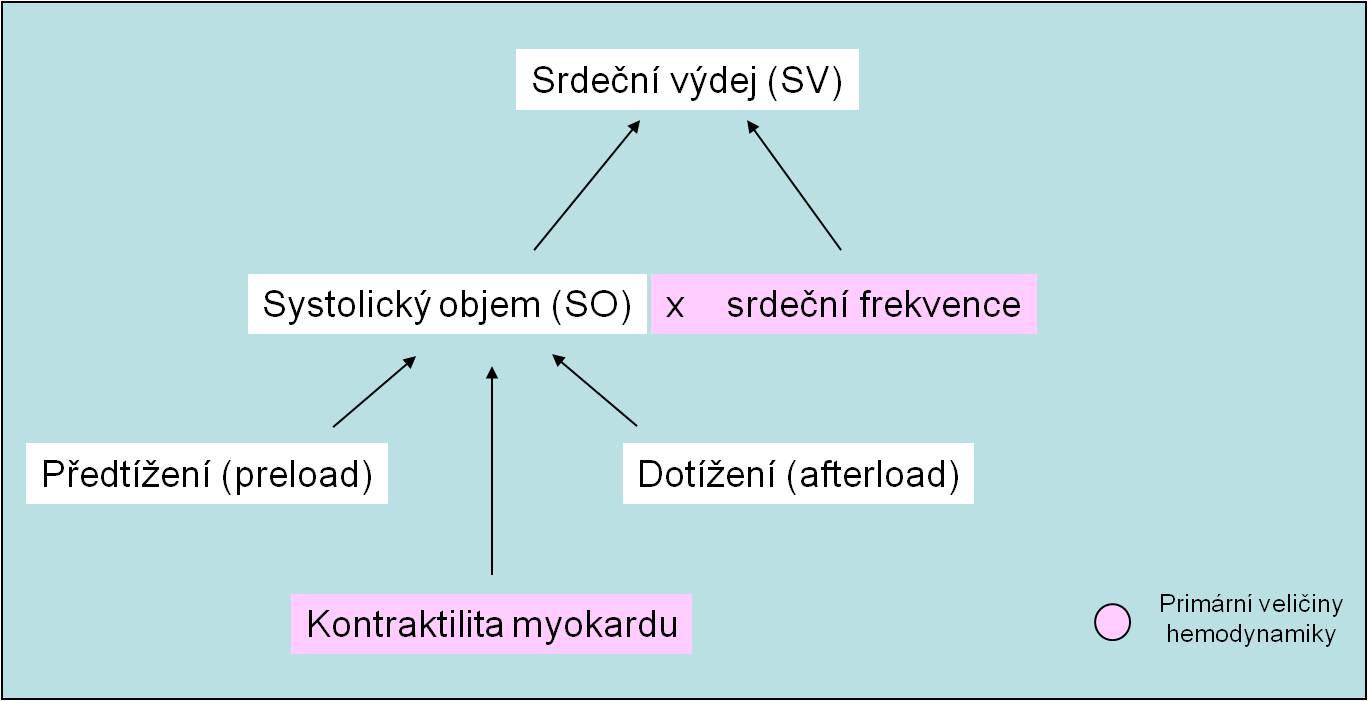
Obr. 1. Determinanty srdečního výdeje (pozor na alternativní značení veličin českými zkratkami na obrázku).
Léčebné ovlivnění těchto parametrů může být kritické pro osud pacienta. Optimalizace předtížení a dotížení na základě změření náležitých klinických ukazatelů, zlepšení kontraktility a zvýšení srdeční frekvence zlepšují srdeční výdej a vyhlíky pacienta na přežití.
Tepový objem
Jak již bylo řečeno výše, jde o objem krve vypuzený z komory během jednoho srdečního stahu a je určen předtížením, kontraktilitou a dotížením (viz obrázek).
Měření tepového objemu
- Neinvazivní techniky
- Echokardiografie
- Měření je založeno na dopplerovském měření rychlosti krevního proudu v aortě. Při vyladění se záznamem EKG lze změřit akceleraci krve po dobu ejekční periody, a tak získat informaci o kontraktilitě komory. Transesofageální echokardiografií, kdy zdroj vln je umístěn v jícnu a snímač na hrudníku, lze potom získat přehled o dynamice pohybu krve v srdečních dutinách a o stavu chlopní.
- Impedanční kardiografie (zpracováno v kapitole Diagnostika oběhu: Impedanční kardiografie)
- Citlivost a přesnost neinvazivní bioimpedanční kardiografie je podle literárních zdrojů srovnatelná s invazivními termodilučními technikami. Na rozdíl od nich poskytuje řadu dalších vysoce potřebných klinických údajů.
- Echokardiografie
- Invazivní techniky
- Termodiluční přístup
- Je založen na podání definovaného množství (10 ml) roztoku o známé teplotě (nižší než má krev) do pravé síně. Tepelné čidlo umístěné za komorou v a. pulmonalis monitoruje změny teploty krve v čase. Z termodiluční křivky je možno určit srdeční výdej. Podobné výsledky poskytuje podání barevně anebo jinak značeného roztoku.
- Fickova metoda
- Fickova metoda měření srdečního výdeje je založena na stanovení úbytku kyslíku (VO2) (ml) ve vdechované směsi za jednotku času (tedy ml O2/min). Pokud zároveň známe arteriovenózní diferenci kyslíku a-vO2 (= CaO2 – CvO2) (ml/l), lze odvodit, kolik kyslíku krev přijala za jednotku času při jejím průchodu plícemi, a odtud vypočítat srdeční výdej (ml/min). Velikost CO se počítá podle Fickova vzorce: CO (ml/min) = [VO2/(CaO2 – CvO2)]. Modifikovaná Fickova metoda se opírá o stanovení částečného zpětného vdechování CO2 a arteriovenózní diferenci CO2.
- Termodiluční přístup
Moderní semiinvazivní techniky
- Systém PiCCO (pulse contour cardiac output)
- Pracuje s centrálním žilním katétrem (je místem aplikace indikátoru – termodilučního roztoku) a arteriálním katétrem zavedeném cestou a. femoralis, a. brachialis nebo jiné periferní arterie (je místem registrace termodiluční křivky za plicním oběhem – namísto sondy v plicnici, která by dovolila stanovit pouze CO). PiCCO kombinuje přerušovaná termodiluční měření registrovaná transpulmonálně s kontinuálním monitorováním hemodynamiky prostřednictvím analýzy tvaru tepové křivky. Srdeční výdej se tedy měří intermitentně stanovením transpulmonální termodiluce (kalibrace, popř. rekalibrace prováděná nejméně každých 6 hodin) a v meziobdobích kontinuálně z monitorované tepové křivky, která se kalibruje podle termodilučních měření.
- Systém dovoluje hodnotit činnost srdce (CO, kontraktilitu) a zároveň objemové poměry v cirkulaci (hydrataci – dehydrataci) a v jejích vybraných oddílech (objemy krve v plicích a v hrudníku, plicní edém, popř. snížený žilní návrat – dehydrataci). Lze jím sledovat účinek terapie, např. farmakologické stimulace inotropie.
- Na rozdíl od měření pulmonálního arteriálního okluzního tlaku (PAOP, pulmonary artery occlusion pressure) anebo tlaku v zaklínění (PAWP, pulmonary artery wedge pressure) nejsou výsledky měření ovlivňovány intratorakálním tlakem anebo kontraktilitou myokardu.
- Systém umožňuje:
- Měřit srdeční výdej (CO), dopočítat srdeční index (CI, normální hodnota 3 – 5 l/min/m2);
- Dopočítat veličiny, jako jsou globální ejekční frakce (GEF) a index srdeční funkce (CFI, normální hodnota 4,5 – 6,5 l/min), které jsou ukazateli kontraktility (inotropie);
- Měřit end-diastolické objemy jednotlivých srdečních oddílů (síní i komor);
- Dopočítat globální end-systolický objem všech srdečních oddílů (GEDV, global end-diastolic volume), který determinuje předtížení (preload) celého srdce;
- Dopočítat objem extravaskulární tekutiny v plicích, a tedy posoudit míru plicního edému (EVLW, extravascular lung water, normální hodnoty 3 – 10 ml/kg, vysoce rizikové hodnoty nad 14 ml/kg);
- Dopočítat objem krve v celém hrudníku (ITBV, intrathoracic blood volume, normální hodnoty ITBV vyjádřené jako index ITBVI jsou 850 – 1000 ml/m2; jde o ukazatel celkového tělesného objemu vody);
- Variace systolického volumu (SVV) anebo variace pulzního tlaku (PPV; pulse pressure variation) - udávají průměrný procentuální rozdíl pulsových tlaků mezi jednotlivými nejvyššími a nejnižšími pulzovými tlaky/systolickými objemy vypuzenými v posledních 30 s. Jde o ukazatel celkového tělesného objemu vody; hodnoty do 10 % jsou normální, hodnoty > 15 % jsou známkou sníženého žilního návratu (dehydratace) – při takových hodnotách lze očekávat příznivou odpověď na podání tekutin.
- Dopočítat systémovou cévní periferní rezistenci (SVR; systemic vascular resistance) – odpor systémového krevního řečiště (dotížení levé komory, levostranný afterload) (normální hodnota SVR: 800 – 1200 dyn x s/cm5).

Obr. 2. Hemodynamický monitor systému PiCCO. Obrázek laskavě poskytl MUDr. Radovan Uvízl, Klinika anesteziologie, resuscitace a intenzivní medicíny LF UP a FN v Olomouci (viz příspěvek Intenzivní medicína v terapii akutního selhání cirkulace).
- Systém LiDCO
- Je variantou předchozího termodilučního systému. Ke kalibraci využívá podání LiCl (do vény) a jeho detekce v arteriální krvi.

Obr. 3. Hemodynamický monitor LiDCO. Obrázek laskavě poskytl MUDr. Radovan Uvízl, Klinika anesteziologie, resuscitace a intenzivní medicíny LF UP a FN v Olomouci (viz příspěvek Intenzivní medicína v terapii akutního selhání cirkulace).
1.faktor: PŘEDTÍŽENÍ (preload)
Předtížení je zátěž, kterou stěna komory vyrovnává svým napětím na konci diastoly. Velikost je především dána end-diastolickou náplní komor, tedy:
- Žilním návratem (periferní determinanta);
- Relaxací a roztažením komor (srdeční determinanta);
- Předchozím vyprázdněním srdce;
- Kontrakcí síní.
Žilní návrat je při stálém periferním odporu primárně determinován středním systémovým plnicím tlakem, tj. především (viz následující obrázek):
- Objemem krve v cirkulaci;
- Napětím velkých kapacitních žil.
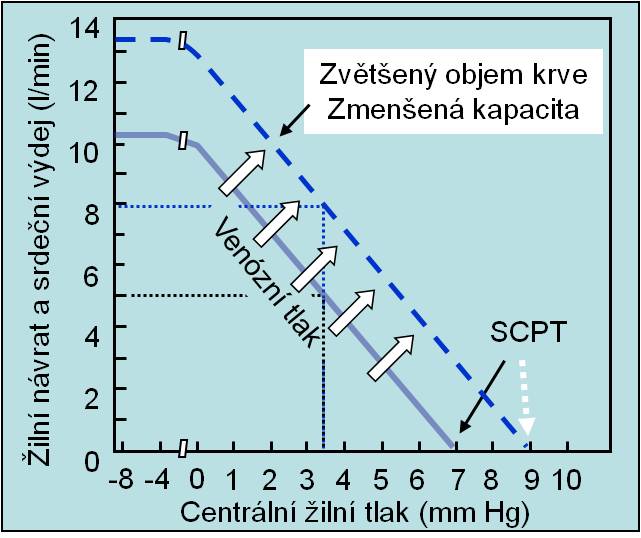
Obr. 4. Graf žilního návratu (venózní funkční křivka) při měnícím se středním cirkulačním plnicím tlaku (SCPT). SCPT se může měnit změnami objemu krve anebo změnami kapacity cirkulace. Se zvýšením SCPT se zvyšuje venózní tlak a žilní návrat, a tedy i srdeční výdej. Snížení SCPT má opačný efekt. Plató vlevo na křivkách je způsobeno kolapsem vén při poklesu žilního tlaku pod tlak v hrudníku. (Podle AC Guyton a JE Hall: Textbook of Medical Physiology, 11th Ed. Elsevier/Saunders, Philadeplhia. 2006.). Schéma je možno zhlédnout i v animované podobě.
Žilní návrat je ovšem rovněž závislý na periferním odporu (viz následující obrázek):
- Při poklesu periferního odporu se zvyšuje;
- Při růstu periferního odporu se snižuje.

Obr. 5. Graf žilního návratu (venózní funkční křivka) při změnách celkového periferního odporu. Se snížením periferního odporu se souběžně zvyšují venózní tlak a žilní návrat, a také srdeční výdej, zatímco střední cirkulační plnicí tlak (SCPT) se nemění. Zvýšení periferní rezistence má opačný efekt. Plató vlevo na křivkách je způsobeno kolapsem vén při poklesu žilního tlaku pod tlak v hrudníku. (Podle AC Guyton a JE Hall: Textbook of Medical Physiology, 11th Ed. Elsevier/Saunders, Philadeplhia. 2006.). Schéma je možno zhlédnout i v animované podobě.
V souladu s právě uvedenými skutečnostmi je třeba brát v úvahu, že žilní návrat je výrazně ovlivněn distribucí krve, např.:
- Změnou tělesné polohy;
- Činností svalové pumpy;
- Tlakem v hrudníku (exspirace; ale zejména Valsalvův manévr – kašel, křeče, umělé dýchání spozitivním tlakem - stlačují cévy a ztěžují návrat k srdci) atd.
Relaxace a roztažení komory za diastoly probíhá následovně:
- Relaxace je aktivní děj, který je ve významné míře určen rychlostí odstranění (sekvestrací) Ca2+ iontů ze sarkoplazmy po kontrakci. Odstranění Ca2+ vyžaduje ATP. Proto výrazně trpí při hypoxii a toxických vlivech. Schopnost aktivní relaxace označujeme termínem lusitropie. Normálně k relaxaci komor dochází v časné, izovolumové fázi diastoly, tj. v období mezi uzavřením aortální anebo pulmonální chlopně a otevřením atrioventrikulárních chlopní. Aktivní relaxace odpovídá za rychlý pokles tlaku v komorách, a tím za jejich počáteční rychlé plnění (fáze rychlého plnění komor).
- V další fázi diastoly se tlaky v síních a komorách vyrovnávají a komory musí na přítok krve odpovídat dostatečnou pasívní poddajností (fáze pomalého plnění komor). Tuto fázi určují fyzikální vlastnosti (tuhost) komory. Je hlavně dána histologickým složením komorových stěn. Poddajnost snižuje hypertrofie (např. při hypertenzi, vadách chlopní, remodelaci), myopatie, přítomnost vaziva (např. fibróza po ischémii) anebo infiltrace komory (amyloidóza, hemochromatóza).
- Diastolické plnění je alterováno při tachykardii anebo tachyarytmiích.
- Relaxaci, poddajnost a plnění také zhoršují mechanické překážky:
- Uvnitř srdce (vady chlopní, remodelace komory, myopatie);
- Vně srdce (tamponáda, změny perikardu).
Lusitropii fyziologicky zvyšuje sympatický vegetativní systém. Přímo působí na svalová vlákna myokardu cestou β-receptorů. Jejich stimulace mimo jiné vede k aktivaci c-AMP-dependentní proteinkinázy A, která fosforyluje protein sarkoplazmatického retikula fosfolamban. Aktivovaný fosfolamban přestává blokovat kalcium-dependentní ATPázu sarkoplazmatického retikula, která potom zvýšeně transportuje Ca2+ zpět do cisteren, a tím napomáhá relaxaci. Zkracuje diastolu.
Porucha lusitropie anebo poddajnosti komory se klinicky manifestuje jako diastolická dysfunkce nebo diastolické selhání.
Pokud jde o kontrakci síní, její význam zejména narůstá při koncentrické hypertrofii komor. Příspěvek síňové kontrakce na druhé straně může být hrubě narušen anebo dokonce chybět při síňové fibrilaci, při poruchách kontraktility síní, atrioventrikulární disociaci anebo při zkrácení, nebo prodloužení intervalu P-R.
Měření předtížení
- V klinické praxi nejčastěji posuzujeme preload podle hodnoty CŽT (preload pravé komory). O předtížení levé komory může informovat tlak v zaklínění (PAWP, pulmonary artery wedge pressure). (Měření PAWP musí být přerušované, nikoliv souvislé, jinak hrozí infarkt plic.) V obou případech jde o parametry tlakové.
- Echokardiografie poskytuje jiný údaj o předtížení – end-diastolický objem pravé komory. V tomto případě jde o parametr objemový.
- Největší citlivost a specifičnost mají ukazatele PiCCO (pulse contour cardiac output, viz výše) informující o objemech krve:
- Ve všech čtyřech dutinách srdce na konci diastoly (GEDV, global enddiastolic volume);
- Jak ve všech čtyřech dutinách srdce na konci diastoly, tak v plicních cévách (ITBV, intrathoracic blood volume). I zde jde o parametry objemové.
- O předtížení vypovídá také variace tepového objemu (SVV, stroke volume variation). Tento dynamický (časově proměnný) parametr lze měřit pouze u pacientů udržovaných na umělé plicní ventilaci (UPV). Ukazuje, jak se cykly UPV promítají do změn srdečního výdeje. Přírůstek SVV lze interpretovat jako indikátor potřeby navýšit objem tekutiny v cirkulaci.
2.faktor: DOTÍŽENÍ (afterload)
Dotížení je zátěž, kterou komora překonává svým napětím po začátku systoly, aby efektivně vypudila krev do arteriálního stromu. Velikost dotížení určují:
- Síla a rychlost kontrakce komor (srdeční determinanta);
- Odpor, který krvi kladou srdeční ústí, velké arterie a jejich náplň (periferní determinanta).
Srdeční složka dotížení, tedy napětí svalu a tlak vyvinutý v komoře jsou (viz Laplaceův zákon, T = P x r/2l, kde T je napětí (tenze) stěny ohraničující kulovitou dutinu, P tlak v dutině, r její poloměr a l tloušťka její stěny):
- Přímo úměrné objemu (náplni) srdeční dutiny;
- Přímo úměrné poloměru komory:
- Dilatace zvětšuje napětí stěny; srdeční výdej klesá;
- Nepřímo úměrné tloušťce stěny dutiny:
- Hypertrofie zmenšuje napětí stěny; srdeční výdej roste.
Periferní složka dotížení levé komory se uplatňuje tlakem krve nad aortálními chlopněmi. Je dána stupněm naplněním aorty a velkých arterií. Závisí na následujících faktorech:
- Na rychlosti plnění aorty a stromu arterií z komory;
- Na relaxaci stěn plněných arterií;
- Na rychlosti vyprazdňování plněných arterií;
- Odtok krve z arterií je hlavně určen periferním odporem. Akutní zmenšení periferního odporu se bezprostředně projeví nižším dotížením (snížený diastolický a střední tlak v aortě); souběžně se zvyšuje předtížení (rostoucí žilní návrat; část krve ovšem zadrží kapacitní oddíl řečiště). Vliv dotížení ukazuje následující obrázek.
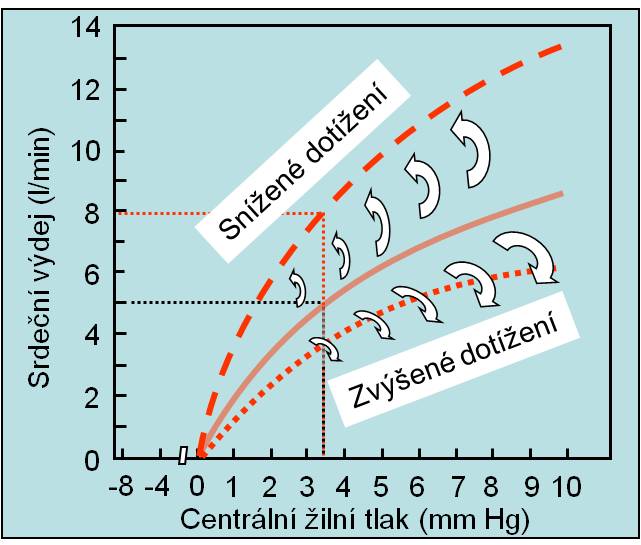
Obr. 6. Změny srdečního výdeje při změnách dotížení. (Podle AC Guyton a JE Hall: Textbook of Medical Physiology, 11th Ed. Elsevier/Saunders, Philadeplhia. 2006.). Schéma je možno zhlédnout i v animované podobě.
Pro klinické účely má význam zejména analýza dotížení levé komory. Závislosti jsou zvláště nápadné u trénovaného anebo u patologicky změněného srdce. Trénované srdce se může lépe vyrovnat s vyšším předtížením i dotížením.
Snížené periferní dotížení usnadňuje ejekci krve z komor. Zvýšené dotížení ztěžuje ejekci. Dotížení proto zejména mění:
- Změny aortálních chlopní;
- Krevní tlak v aortě a v arteriích;
- Změny poddajnosti velkých arterií;
- Konstrikce, nebo dilatace periferních arteriol;
- Arteriovenózní zkraty.
Dotížení dosahuje svého horního limitu, když srdce ani při krajním napětí vláken nedokáže otevřít aortální chlopně a vypudit krev do aorty.
Měření dotížení
- V klinické praxi se jako ukazatel dotížení levé komory hodnotí periferní cévní odpor v systémové cirkulaci (SVR, systemicc vascular resistance). O dotížení pravé komory podobně informuje plicní vaskulární rezistence (PVR).
- Odpor se podobně jako v nauce o elektřině počítá ze znalosti velikosti proudu a tlaku:
- SVR = [(MAP - CVP)/CO] x 80 = (PP/CO) x 80
- PVR = [(MPAP - PAOP)/CO] x 80
- (MAP, střední systémový arteriální tlak; MPAP, střední arteriální tlak v plicnici; CVP, centrální žilní tlak; PAOP, vstupní arteriální tlak v plicnici, pulmonary artery opening pressure; CO, srdeční výdej).
3. faktor: SÍLA A RYCHLOST STAHU MYOKARDU
Mechanická práce srdce se stejně jako u kosterního svalstva manifestuje jednak napětím, jednak zkrácením svalu. Dožení určitého napětí je nezbytné pro vyrovnání zátěže a překonání odporu, proti kterému srdce svým stahem vypuzuje krev. Rychlost a míra zkrácení svalu jsou nepřímo úměrné tomuto napětí. Závislost lze znázornit křivkami zátěž-rychlost.
Na rozdíl od kosterního příčně pruhovaného svalu, jehož síla roste především náborem většího počtu svalových jednotek vybuzených zvýšenou nervovou impulsací, zůstává množství svalových vláken zajišťujících srdeční stahy konstantní. Sílu a rychlost zkrácení srdečního svalového vlákna za takových okolností podle potřeby spoluurčují dva faktory:
- Kontraktilita (inotropie). Popisuje výkon svalového vlákna za různých situací při jeho nezměněné výchozí délce.
- Je dána jednak metabolickým a energetickým stavem svalových vláken, jednak přítomností stimulačních, nebo inhibičních faktorů;
- Určuje úroveň, na které pracuje Frank-Starlingův mechanismus.
- Výchozí délka svalových vláken. Hovoří se o aktivaci svalového vlákna jeho protažením před začátkem stahu.
- Působí i při nezměněné kontraktilitě.
- Popisuje se jako Frank-Starlingův mechanismus (Frank-Starlingův zákon);
- Je dána diastolickým objemem, tedy end-diastolickou náplní srdce.
Kontraktilita (síla stahu vlákna o určité délce)
Na rozdíl od kosterního příčně pruhovaného svalu mají akční potenciály svalových vláken myokardu jasně vyjádřeno ploché vrcholové plató. V jeho průběhu proudí malé množství iontů kalciovými kanály typu L obsaženými v plazmatické membráně do sarkoplasmy, kde vyvolá uvolnění velkého množství iontů Ca2+ z cisteren sarkoplazmatického retikula. Tyto ionty se potom vážou na troponin C, potlačí jeho inhibiční působení na vzájemné interakce myofilament, tzn. aktinu amyosinu, a umožní začátek kontrakce.
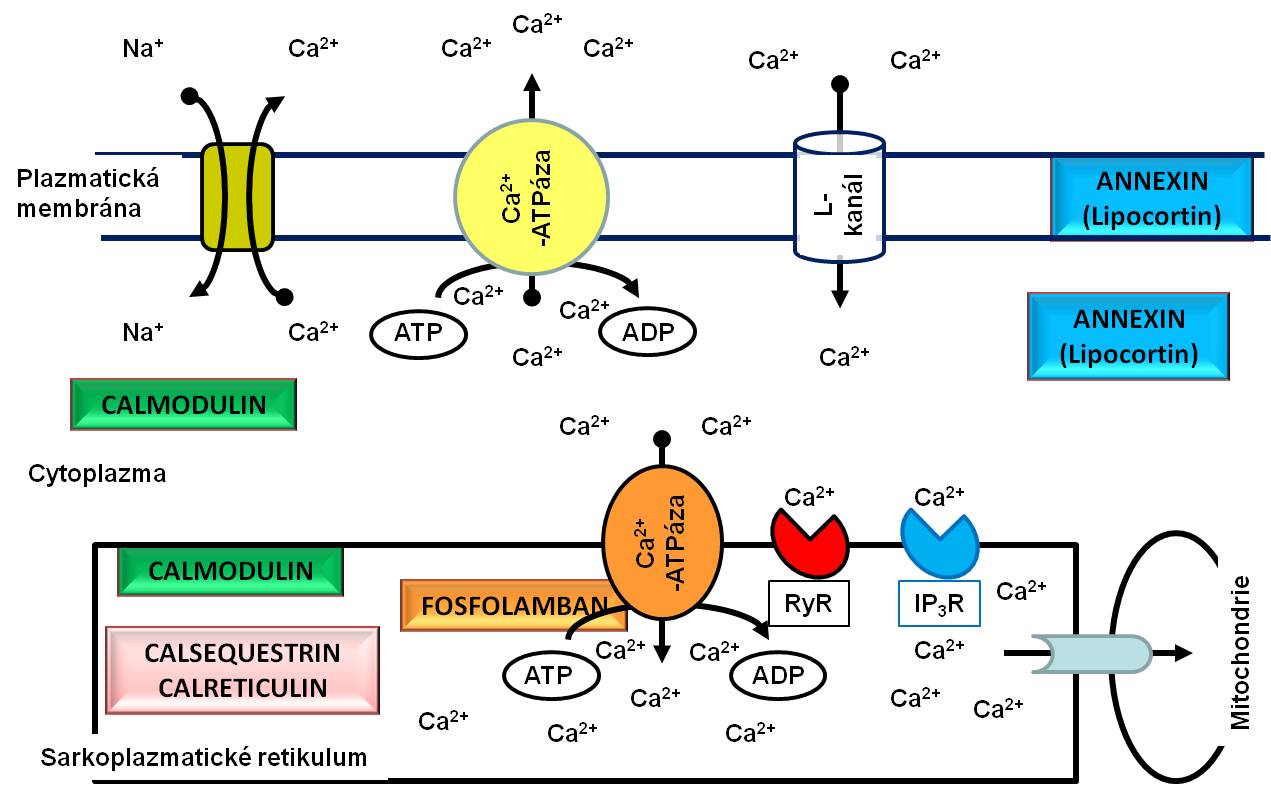
Obr. 7. Dynamika kalciových iontů při svalovém stahu. RyR, ryanodinový receptor; IP3R, receptor inositoltrisfosfátu.
V sarkoplasmě není při svalovém stahu dostatek Ca2+, aby se nasytila všechna vazebná místa na troponinu C pro tyto ionty. Procesy, které zvyšují koncentraci Ca2+ v cytosolu svalového vlákna, zvyšují sílu kontrakce svalu bez ohledu na jeho výchozí délku. To je podstatou ovlivnění kontraktility. Ví se ovšem, že fenomén je poněkud složitější, protože změny koncentrace Ca2+ ho úplně nevysvětlují.
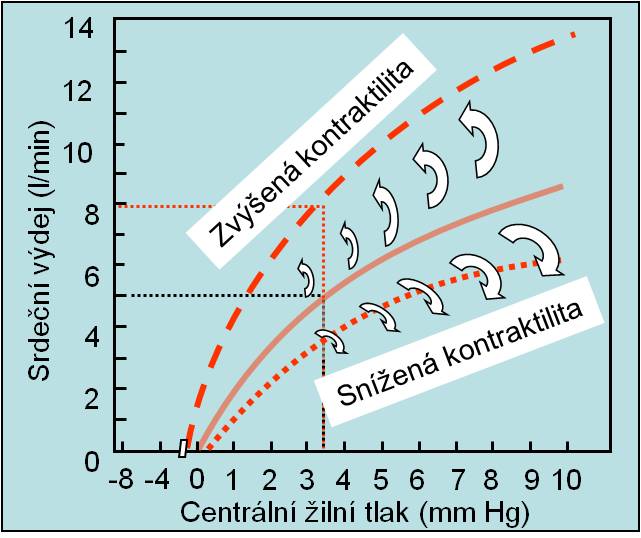
Obr. 8. Změny srdečního výdeje při změnách kontraktility. Křivka srdečního výdeje (kardiální funkční křivka) se při zvýšení kontraktility odklání nahoru a posunuje se doleva. Výsledkem je zvýšený srdeční výdej i při poklesu plnicího žilního tlaku. Snížení kontraktility působí opačně. Dolní limit poklesu kontraktility je dán komorovým tlakem potřebným pro otevření aortálních chlopní. (Podle AC Guyton a JE Hall: Textbook of Medical Physiology, 11th Ed. Elsevier/Saunders, Philadeplhia. 2006.). Schéma je možno zhlédnout i v animované podobě.
Kontraktilita (obr. 1, obr. 8) není závislá na předtížení anebo dotížení. Kontraktilitu ovlivňují:
- Metabolické vlivy (hypoxie, hyperkapnie, acidóza, iontové dysbalance; stav koronárních cév);
- Nervové a hormonální vlivy (sympatikus, parasymaptikus, cirkulující hormony);
- Poddajnost myokardu
- Objem svalové hmoty myokardu;
- Frekvence (frekvenční efekt);
- Farmaka (kardiotonika, kardiodepresiva).
Sympatický vegetativní systém přímo působí na vlákna svalová myokardu a ovlivňuje kontraktilitu cestou β-receptorů. Jejich stimulace mimo jiné vede k aktivaci c-AMP dependentní proteinkinázy A, která fosforyluje Ca2+ kanály typu L sarkolemy, a tím napomáhá vstupu iontů Ca2+ z extracelulárního prostoru do vláken. Navýšením kontraktility roste rychlost a síla stahu, zkracuje se systola, zvětšuje se systolický volum a zvyšuje se rychlost krve vypuzované do aorty.
Měření kontraktility
- Kvantitativní odhad kontraktility je možno provést ze strmosti vzestupu pulsové křivky při přímém měření arteriálního tlaku.
- Jiným prakticky používaným ukazatelem je výkon komory, tzn. hodnoty tepové práce (SW, stroke work) levé komory (LVSW) anebo pravé komory (RVSW) za časovou jednotku:
- LVSW = SV x (MAP – PAOP) x 0,0136
- RVSW = SV x (MPAP – CVP) x 0,0136
- (SV, systolický neboli tepový volum; MAP, střední arteriální tlak; MPAP, střední arteriální tlak v plicnici; PAOP, vstupní tlak v a. pulmonalis, pulmonary artery opening pressure; CVP, centrální žilní tlak).
- Důležitou informaci o kontraktilitě rovněž podává globální ejekční frakce (GEF) a index srdeční funkce (CFI). Zjišťují se z měření systémem PiCCO (viz výše).
Frank-Starlingův mechanismus (aktivace určená protažením)
S prodlužující se výchozí délkou svalového vlákna se v určitých mezích optimalizuje prostor pro vzájemnou interakci obou hlavních druhů myofilament, tzn. aktinu a myosinu. Roste citlivost myofilament k Ca2+, a klesá koncentrace Ca2+, které je zapotřebí pro dosažení 50 % maximálního napětí vlákna. Proto tlak vyvinutý komorou při systole je tím větší, čím více jsou protažena svalová vlákna svalu před začátkem stahu (při nezměněné kontraktilitě). Systolický volum tak roste tím více, čím více se srdce naplnilo během diastoly (viz obrázek výše). Jde o aktivitu srdce závislou na předtížení.
Frank-Starlingův mechanismus zajišťuje, že srdce vypumpuje – při nezměněné kontraktilitě – stejné množství tekutiny, jaké do něho přiteče. Při náhlé změně žilního návratu anebo vyprazdňování srdce se tak během několika stahů ustálí nová hemodynamická rovnováha.
4. faktor: SRDEČNÍ FREKVENCE
S růstem srdeční frekvence dosahuje srdeční výdej plató. Růst frekvence nad oblast plató (> 160/min) má negativní dopad. Je to patrné z uvedeného grafu. Růst frekvence jde na úkor většího zkracování diastoly než systoly. Komory se pak v průběhu krátké diastoly nestačí naplnit.
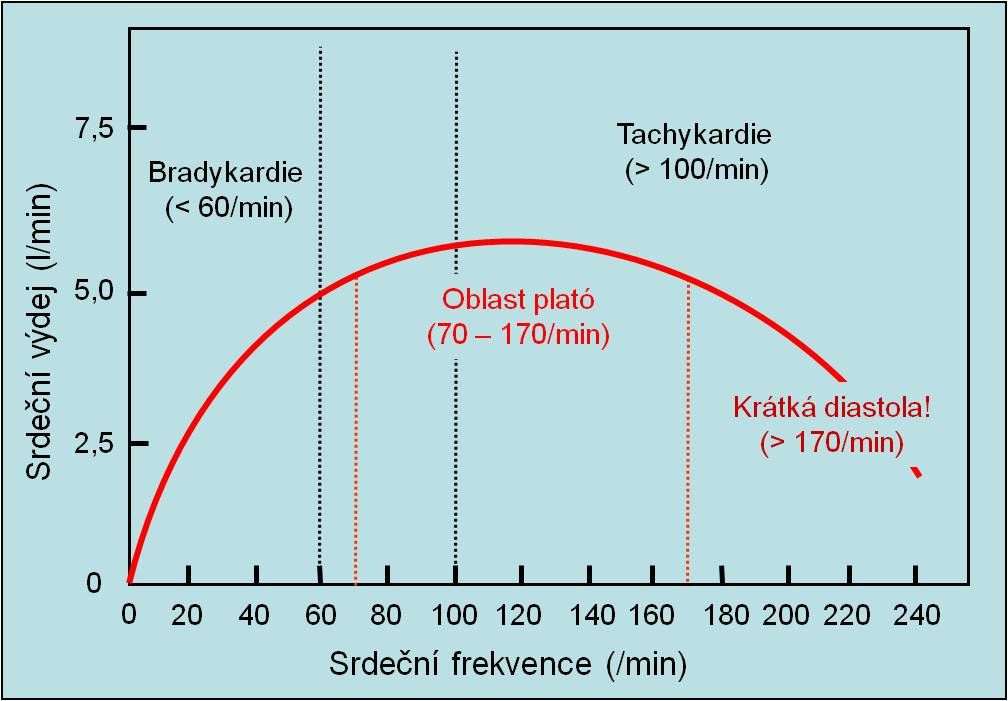
Obr. 9. Změny srdečního výdeje se změnami srdeční frekvence. (Podle AC Guyton a JE Hall: Textbook of Medical Physiology, 11th Ed. Elsevier/Saunders, Philadeplhia. 2006.)
Srdeční selhání
Definice srdečního selhání
Srdeční nedostatečnost nebo srdeční selhání je stav, kdy srdce není schopno udržet dostatečný srdeční výdej pro pokrytí potřeb tkání anebo pokrývá tyto potřeby jen za cenu trvalého zvýšení konečného komorového diastolického objemu.
Při latentním selhání srdce stačí pokrýt nároky v klidu a srdeční nedostatečnost se manifestuje pouze při námaze.
K selhání srdce nedochází vždy tak, že výkon srdce (srdeční výdej) postupně klesá. Existuje také srdeční selhání rozvíjející se po období zvýšeného, nebo dokonce vystupňovaného srdečního výdeje (viz níže odstavec Srdeční selhání s nízkým srdečním výdejem a srdeční selhání s vysokým srdečním výdejem).
Srdeční selhání je provázeno abnormálním koloběhem kalciových iontů v sarkoplazmě příčně pruhovaných vláken myokardu a abnormální interakcí Ca2+ s kontraktilními proteiny:
- Bazální (diastolická) cytoplazmatická koncentrace Ca2+ je zvýšena;
- Vrcholová (systolická) koncentrace Ca2+ je nižší;
- Opětovný pokles cytoplazmatické koncentrace Ca2+ po kontrakci vlákna je pomalý.
Obecné příčiny srdečního selhání
Nejčastější příčinou selhání srdce jako pumpy je poškození myokardu. V takových případech je synonymem srdečního selhání selhání myokardu. Selhání myokardu musíme odlišovat od nedostatečného srdečního výdeje (selhání cirkulace) způsobeného náhlou insuficiencí chlopně anebo náhlou rupturou papilárního svalu např. při endokarditidě anebo při infarktu myokardu, kdy je dosud normální myokard vystaven náhlému přetížení, které přesahuje jeho momentální rezervní funkční kapacitu. Podobně je tomu při objemovém přetížení vyvolaném náhlým zvýšením žilního návratu, nebo při tlakovém přetížení vyvolaném hypertenzní krizí nebo masivní embolizací do plicnice apod. Stejně tak si myokard zachovává své funkce i při selhání cirkulace vyvolaném srdeční tamponádou (tekutinou v perikardu, změnou perikardu).
Jako srdeční selhání rozhodně nelze označovat stavy, kdy je nedostatečný srdeční výdej způsobený nízkým žilním návratem, např. při krvácení anebo hypovolémii. Srdečním selháním není ani městnání vyvolané přílišným hromaděním tekutin, jaké např. provází selhání ledvin; hromadění tekutin v těle však následně může vést k objemovému přetížení, a selhání srdce.
Akutní a chronické selhání
Akutní selhání se vyvíjí nečekaně, nejčastěji z ischemických příčin anebo na podkladě náhlého tlakového nebo objemového přetížení srdce. V takových případech nejčastěji jde o systolické selhání s nízkým srdečním výdejem a s následnou systémovou arteriální hypotenzí. Akutní srdeční selhání není provázeno periferními otoky.
Naproti tomu při chronickém selhání systémový arteriální tlak dlouho zůstává zachován z důvodu masivní retence tekutin ledvinami, přeplnění cirkulace a využití srdeční rezervy umožněné Frank-Starlingovým mechanismem. Na rozdíl od akutního selhání jsou pro chronické selhání charakteristické rozsáhlé periferní otoky.
Koncentrická a excentrická hypertrofie
Myokard odpovídá na chronicky zvýšenou zátěž hypertrofií. Při chronickém tlakovém zatížení se vyvíjí koncentrická hypertrofie (typicky při arteriální hypertenzi). Hmotnost komory roste, protože srdeční stěna se ztlušťuje, takže se v ní v soluladu s Laplaceovým zákonem zmenšuje napětí. Koncentricky hypertrofovaná komora může roky plnit své funkce, než nakonec dilatuje. Variantou je koncentrická remodelace, kdy se tloušťka komory rovněž relativně zvětšuje v odpověď na tlakové zatížení, ale hmotnost komory neroste a zmenšuje se volum dutiny, takže je nízký systolický výdej (malá náplň, „objemové podtížení“).
Při chronickém objemovém přetížení vzniká excentrická hypertrofie (dilatace) komory. Typicky provází nedomykavost srdečních anebo aortálních chlopní. Hmotnost komory roste, stěna se ztlušťuje úměrně dilataci komory, takže alespoň zpočátku zůstává poměr mezi tloušťkou stěny a velikostí dutiny zachován. Později dochází k dilataci na úkor tloušťky stěny a sférické remodelaci komory. Úbytek tloušťky je provázen zvyšováním napětí ve stěně a vznikem bludného kruhu.
Pravostranné a levostranné selhání (selhání pravého srdce a selhání levého srdce)
Krev z pravé komory proudí do levého srdce přes plíce, a tak je namístě rozlišovat levostranné a pravostranné srdeční selhání. Jejich symptomy se navzájem liší. Přesto mají obě tyto formy selhání některé symptomy společné. Tak při selhání pravé komory trpí plnění levé komory, a proto vázne srdeční výdej do systémového oběhu podobně, jako když selhává levá komora. Na druhé straně je selhání levé komory provázeno plicní hypertenzní, která sekundárně může vést k alteraci až selhání pravé komory.
Také je vhodné mít na paměti, že svalovina levé komory a svalovina pravé komory od sebe nejsou navzájem odděleny. Srdeční svalovina spojitě okružuje dutiny obou komor. Obě komory navíc sdílejí společné septum. Proto poruchy svaloviny jedné části myokardu ovlivňují funkci ostatních částí srdečního svalu.
Selhání dozadu (zpětné selhání; backward) a selhání dopředu (dopředné selhání; forward)
Odděleně lze hovořit o selhání dozadu a selhání dopředu pouze z didaktických důvodů. Ve skutečnosti jde o dvě stránky téhož jevu, které se nemohou vyskytovat jedna bez druhé. Selhání se vždy projeví nějakými následky jak před selhávajícím oddílem (selhání dozadu, zpětné selhání, tzn. proti proudu krve; backward, nebo také upstrem), tak následky za ním (selhání dopředu, po proudu krve; forward, downstream). Obě formy se tak spolu vždy vyskytují současně.
Termín selhání dozadu (backward) zdůrazňuje fakt, že před selhávajícím srdečním oddílem, tedy proti proudu, dochází ke stagnaci krve, kterou postižená komora nemůže přečerpat dál.
Akutní následky levostranného selhání, které se projevují dozadu, mohou být fatální. Typicky k tomu dochází při akutním infarktu levé komory. Pravá komora, která není postižena, pumpuje krev s normální intenzitou, a tak dochází k městnání krve v plicním oběhu před selhávající levou komorou. Vyvíjí se plicní hypertenze s vysokým rizikem vzniku akutního plicního edému. Po překonání bezpečnostních faktorů (animace – bezpečnostní faktory) se rozvíjí plicní edém – zprvu intersticiální, později alveolární. Edém se bude manifestovat příznaky dyspnoe, asthma cardiale anebo smrtícím akutním plicním edémem.
Při chronickém levostranném selhání vyvolává přetrvávající plicní hypertenze hypertrofii pravé komory, která se nakonec může dilatovat a selže. Selhávající pravá komora přestane udržovat plicní hypertenzi, takže pokud jde o respirační příznaky, pacientovi se paradoxně uleví. Nicméně dosavadní symptomy levostranného selhání se zároveň rozšíří o periferní známky chronického pravostranného selhání.
Akutní pravostranné selhání směrem dozadu způsobí stagnaci krve v žilách systémového oběhu. Projeví se distenzí systémových vén a městnavou hepatomegalií a splenomegalií. Typicky to můžeme vidět při rozsáhlejší plicní embolii. Akutní pravostranné selhání neprovázejí periferní otoky. Ty se nejdříve objeví až po několika desítkách hodin anebo - typicky - až po několika dnech. Souvisejí s ději, které se rozvinou ve směru dopředu, tj. po proudu krve, za selhávajícím oddílem.
Akutní následky levostranného selhání, které se projevují dopředu, tj. po proudu krve, jsou vyvolány nízkým srdečním výdejem. Při jeho kritickém poklesu může dojít k šoku a ke smrti. Kompenzace nízkého srdečního výdeje se účastní celá řada mechanismů. Hlavní jsou následující:
- Pokles systémového arteriálního tlaku registrovaný baroreceptory vede k aktivaci sympatického systému, který kromě svých kardiálních účinků akutně zprostředkuje redistribuci a centralizaci oběhu.
- V další fázi podpoří centralizaci oběhu systém renin-angiotenzin, aktivovaný v hypoperfundovaných ledvinách.
- Zásadní kompenzační účinek sympatiku a renin-angiotenzinového systému dále spočívá v přetrvávající vystupňované retenci sodných iontů a vody v ledvinách. Tímto působením vzrůstá střední cirkulační plnicí tlak, a zvětšuje se diastolické plnění srdce. Zvýšený preload Frank-Starlingovým mechanismem stimuluje srdeční výdej. Pokud se výkon srdce zlepší tak, že hypoperfúze ledvin odezní, celý tento mechanismus se vypne.
- Natrium a vodu-šetřící účinky sympatiku a angiotenzinu II mohou být dále podpořeny zvýšenou sekrecí aldosteronu a antidiuretického hormonu.
- Zároveň se vzrůstem cirkulačního plnicího tlaku se zvyšuje střední hydrostatický tlak v krevních kapilárách, který postupně může převážit nad onkotickými silami a vést k periferním otokům.
Dopředná charakteristika (po proudu) pravostranného srdečního selhání se kryje s popisem levostranného dopředného selhání. Plícemi přichází ze selhávající pravé komory do levé komory zmenšený proud krve, takže klesá srdeční výdej do systémového oběhu. Nemusí jít jen o akutní stavy, jak to ilustruje výše vzpomenutá plicní embolie, ale i o chronická onemocnění, např. plicní emfyzém provázený cor pulmonale. Dopředné pravostranné selhání rovněž může akutně vést k šoku anebo k postupnému zvyšování středního cirkulačního plnicího tlaku a k vývoji městnání v malém anebo systémovém oběhu, jak bylo popsáno v předchozím odstavci.
Zvyšování středního cirkulačního plnicího tlaku hypoperfundovanými ledvinami je jedním z klíčových dějů, které při poškození srdce mohou pomocí Frank-Starlingova mechanismu zajistit potřebný návrat srdečního výdeje nad kritickou úroveň. Pokud však je poškození srdce těžké a potřebného prahového srdečního výdeje, který by dokázal uspokojit požadavky ledvin, se nedosáhne, způsobí narůstající retence tekutin a plnění srdce překročení optimální délky sarkomer (body C a D na obrázku). Další zvyšování předtížení už nepovede ke zvyšování, ale naopak ke snižování srdečního výdeje, a fatálnímu konci (body E a F na obrázku). V této fázi, kdy už také bývají přítomny masivní otoky, je naopak nezbytné zabránit další akumulaci tekutin diuretiky. Rozvíjející se městnavé selhání signalizují zvýšené koncentrace natriuretických peptidů v plazmě, jejichž stanovení se využívá klinicky.
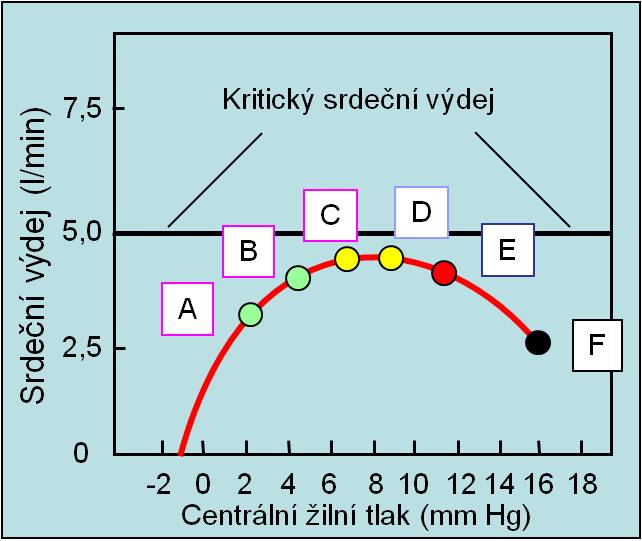
Obr. 10. Ilustrace neefektivního zvyšování středního cirkulačního plnicího tlaku, které při těžkém poškození srdce vedlo k výraznému městnání a ani po dosažení kritického plnění pravé síně nedokázalo Frank-Starlingovým mechanismem navýšit srdeční výdej na dostatečnou úroveň. (Podle AC Guyton a JE Hall: Textbook of Medical Physiology, 11th Ed. Elsevier/Saunders, Philadeplhia. 2006.)
Systolické a diastolické selhání
Při systolickém selhání spočívá hlavní porucha v neschopnosti komory vypudit dostatečné množství krve. Její schopnost plnit se je zachována.
Při diastolickém selhání spočívá hlavní porucha v neschopnosti komory přijmout potřebnou náplň. Často se obě poruchy kombinují.
Primárními příčinami systolického selhání mohou být:
- Ischemické poškození;
- Primární (idiopatické) kardiomyopatie;
- Arteriální hypertenze;
- Chlopňové vady.
Při systolickém selhání je porušena kontraktilita, oslabena kontrakce, zhoršeno vyprazdňování komory. Hlavní funkční charakteristikou systolického selhání je snížení polohy a zploštění křivky kontraktility. Jak ukazuje křivka c na obrázku 11, komora neodpovídá na zvýšenou diastolickou náplň (protažení vláken) potřebnými přírůstky výkonu, tj. srdečního výdeje. Průběh plnění srdce je normální (není přítomno diastolické selhání), ale snižuje se ejekční frakce, takže se zvyšuje end-diastolický objem a tlak. Systolické selhání se zpravidla manifestuje jako selhání s nízkým srdečním výdejem. Srdeční výdej je nedostatečný. Obojí ukazují následující obrázky 11 a 12 (křivka Dilatace na obr. 12).
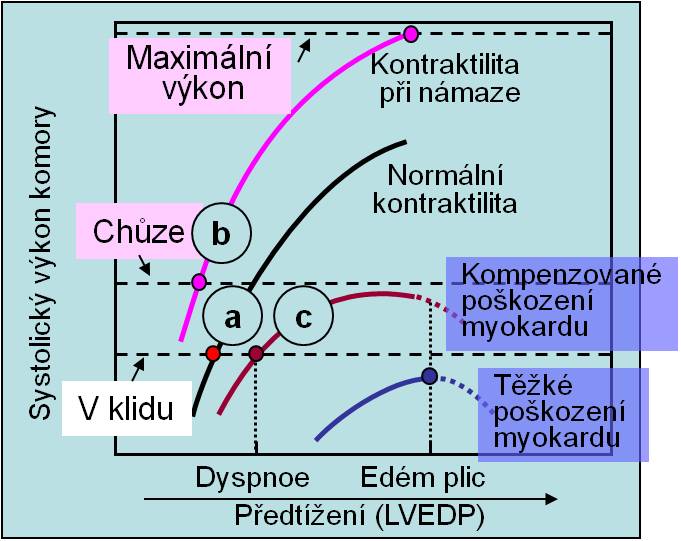
Obr. 11. Změny předtížení a srdeční výdej při různých úrovních kontraktility. Křivka a: Normální stav (klidová kontraktilita, klidový srdeční výdej = 5 l/min). Křivka b: Při námaze se zvyšuje kontraktilita; end-diastolický objem levé komory (LVEDP) se snižuje. Podobně rovněž při koncentrické hypertrofii zůstávají systolické funkce zachovány, ale vázne dilatace a end-diastolický objem je nižší; následkem může být nízký srdeční výdej (diastolické selhání; na obrázku neukázáno). Křivka c: Začínající systolické selhání. Dostatečný srdeční výdej v klidu se udržuje za cenu vyššího end-diastolického objemu (Frank-Starlingův mechanismus). Chybí rezerva. Křivka d: Těžké systolické selhání. Srdeční výdej nedosahuje ani klidového výdeje, přestože end-diastolický objem je zvýšený. (Podle Harrison´s Principles of Internal Medicine, 15th Ed. McGraw-Hill, New York, 2001.).
Při diastolickém selhání je primárně porušena relaxace (lusitropie) anebo poddajnost komory. Během plnění komory strmě roste nitrokomorový tlak a možný maximální end-diastolický objem je redukovaný. Ejekce náplně z komory probíhá normálně a srdeční výdej zůstává normální (není přítomno systolické selhání; proto se diastolické selhání vyčleňuje jako do jisté míry specifická forma srdečního selhání se zachovanou systolickou funkcí). Jak ukazuje obrázek 9, hlavní funkční charakteristikou diastolického selhání je strmá křivka závislosti diastolického tlaku na diastolické náplni komory (křivka „Hypertrofie“ na obrázku 12).
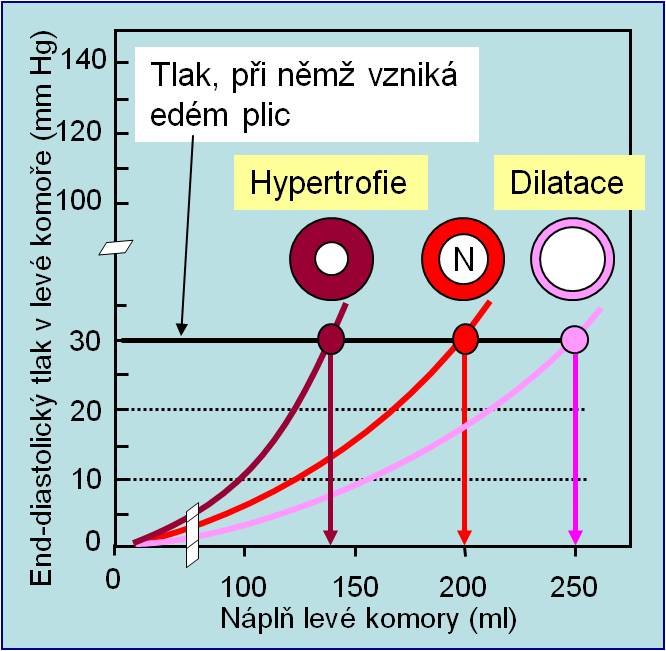
Obr. 12. Vztah mezi end-diastolickým tlakem a end-diastolickým objemem v levé komoře v normálním stavu, při hypertrofii komory a při dilataci komory. Normální levá komora (N) může pojmout 200 ml krve, než v ní end-diastolický tlak kriticky stoupne na 30 mm Hg (vznik plicního edému). End-diastolický tlak v koncentricky hypertrofické komoře dosáhne kritické hodnoty už při náplni 130 – 140 ml. Komora je mnohem méně poddajná. Jde o tzv. diastolické selhání, protože systolické funkce (systolický volum) zůstávají v normě. Naproti tomu dilatovaná komora může pojmout více než 250 ml, než nastane kritický vzestup tlaku. Je mnohem více poddajná. Jde o tzv. systolické selhání, protože systolické funkce komory (systolický volum) jsou od začátku redukovány. (Podle Harrison´s Principles of Internal Medicine, 15th Ed. McGraw-Hill, New York, 2001.).
Diastolické selhání je přítomno u nejméně 50 % všech případů chronického srdečního selhání. Jeho prevalence roste s věkem a mezi postiženými mírně převažují ženy. Více jsou postiženi pacienti s hypertenzí (méně trpí systolickým selháním) než pacienti s prodělaným infarktem, tzn. ischemickou srdeční chorobou.
Termín diastolické selhání (stejně jako termín diastolická dysfunkce), by měl zůstat rezervován pro poruchy komorového plnění primárně způsobené postižením relaxace (lusitropie) anebo poddajnosti myokardu. Termín srdeční selhání se zachovanou systolickou funkcí je širší a zahrnuje i stavy, při nichž vázne plnění, aniž byla poškozena relaxace anebo poddajnost samotné stěny komory.
S diastolickým selháním anebo srdečním selháním se zachovanou systolickou funkcí se typicky setkáváme za následujících okolností:
- Přechodná (funkční) diastolická dysfunkce zpravidla provází ischémii.
- K poruše plnění může dojít také při tachykardii, tachyarytmiích, anebo při absencích síňových kontrakcí.
- Chronická diastolická dysfunkce často provází hypertenzivní koncentrickou hypertrofii komory; zřídka vede k systolickému srdečnímu selhání.
- Riziko srdečního selhání je vysoké u hypertrofií spojených s vazivovou přestavbou stěny, jako jsou:
- Hypertrofická kardiomyopatie;
- Restriktivní kardiomyopatie a fibróza (při infiltrativních onemocněních).
- Porucha plnění bez primární poruchy myokardu provází konstriktivní perikarditidu.
- Při současné hypertrofii a dilataci se obě poruchy, systolické a diastolické selhání, kombinují.
Selhání s nízkým srdečním výdejem a selhání s vysokým srdečním výdejem
Normální srdeční výdej kolísá v širokých mezích (3,7 – 6 l/min neboli 2,2 – 3,5 l/min/m2), takže hodnocení podle absolutní hodnoty srdečního výdeje je nespolehlivé. Jak pro selhání s nízkým výdejem, tak pro selhání s vysokým výdejem jsou charakteristické ploché křivky srdečního výdeje v závislosti na diastolickém plnění srdce, a jejich význam pro diferenciální diagnózu je tedy omezený. Pro spolehlivé odlišení obou stavů je zapotřebí přihlédnout k patogenetickým hlediskům.
V pozadí selhání charakterizovaného nízkým srdečním výdejem může být systolické selhání, diastolické selhání, anebo jejich kombinace. Jejich příčiny byly stručně zmíněny výše. Tkáně jsou při nízkém srdečním výdeji nedostatečně zásobeny krví a kyslíkem, a proto z krve odebírají více kyslíku, takže roste kyslíková arteriovenózní diference. K selháním s nízkým srdečním výdejem nepatří selhání cirkulace, ke kterému může dojít následkem sníženého žilního návratu (např. při krvácení, absolutní anebo relativní hypovolémii, nebo obstrukci vén), a jehož příčiny tak leží mimo srdce.
Srdeční selhání, které se vyvine na základě předchozího chronicky zvýšeného srdečního výdeje, předchází hemodynamické období se sníženou periferní rezistencí (hyperkinetická cirkulace). Příklady jsou stavy hypervolémie, chronické hypoxie, hypoxémie, anémie, hypertyreoidismus, arteriovenózní zkraty, Pagetova nemoc, nebo beri-beri. Klinicky se projevují hyperkinetickou cirkulací. Ne zcela doceňovaná je hyperkinetická zátěž srdce při obezitě. Hyperkinetická cirkulace fyziologicky provází těhotenství. Krátkodobě se objevuje např. při horečce, při fyzické práci. Kyslíková arteriovenózní diference při srdečním selhání s vysokým srdečním výdejem může být v závislosti na povaze primární poruchy snížená, normální, nebo zvýšená.
Stav blízký hyperkinetické cirkulaci vytváří insuficience aortální chlopně.
Klinické symptomy srdečního selhání
Celkové nespecifické příznaky zahrnují periferní vazokonstrikci, bledost, chladnou kůži, únavnost, bolesti hlavy a sníženou výkonnost. Mohou být přítomny nechutenství, nausea a příznaky intestinální dyspraxie. Pokud se vyvine cyanóza, nejdříve postihne rty a nehtová lůžka. Při těžkém selhání mohou být přítomny vystupňované známky ischémie centrálního nervového systému, jako jsou poruchy paměti, nespavost, deprese anebo zmatenost.
Kardiální otoky se nejprve symetricky objevují na nejníže položených místech těla, později se vyvíjí ascites a hydrothorax. Městnání bývá provázeno hepatosplenomegalií. Pozdním nálezem je poškození jater a žloutenka.
Puls bývá pravidelný, pulsový tlak může být snížen z důvodu sníženého systolického objemu. Diastolický tlak a centrální venózní tlak bývají zvýšeny při retenci tekutin.
Časným příznakem ze strany respiračního systému je dyspnoe. Nejdříve je vázána na námahu a signalizuje sníženou funkční rezervu. Později je přítomna i v klidu. Pokud se vyvíjí plicní edém, snižuje se poddajnost plic, takže dýchání je spojeno s větším úsilím. Záchvatům noční dyspnoe napomáhá jak útlum respiračního centra, tak snížená stimulace srdce při snížené sympatické aktivitě ve spánku.
Ortopnoe je vystupňovaná dyspnoe. Vzniká z důvodu redistribuce krve, která se v těle v poloze vleže přesunuje z dolních končetin a břišní dutiny do hrudníku, kde zhoršuje plicní edém.
Noční záchvaty ortopnoe mohou mít podobu kardiálního astmatu (asthma cardiale) s kašlem, expektorací řídkého zpěněného sputa a se sípáním způsobeným bronchospasmem.
Zvláštním příznakem je hubnutí, které probíhá pod obrazem kardiální kachexie. Pacienti trpí progresivním úbytkem svalové hmoty a výraznou lipolýzou, což jsou známky terminální kachexie. Na vzniku kardiální kachexie se podílejí:
- Zvýšený metabolismus (hypermetabolismus; stresové hladovění), který je vyvolán:
- Cirkulujícími zánětlivými cytokiny (např. TNF);
- Inzulinovou rezistencí;
- Zvýšenými nároky respiračních svalů na dodávky kyslíku a energie;
- Zvýšenými nároky hypertrofovaného myokardu na dodávky kyslíku a energie.
- Nechutenství a poruchy funkcí gastrointestinálního traktu (intestinální dyspraxie), které jsou vyvolány:
- Poruchami pasáže a vstřebávání způsobenými hypoxií a městnáním;
- Pocity plnosti vyvolanými hepatosplenomegalií a meteorismem;
- Enteropatie provázená sekrecí proteinů do střeva;
- Vedlejšími účinky léků.
Diagnostika poruch a hodnocení výkonnosti srdce
Klidová vyšetření
Známkou systolické (kontraktilní) poruchy levé komory může být zvětšený end-diastolický objem anebo snížená ejekční frakce. Zvýšené dotížení reprezentované zvýšeným end-diastolickým tlakem v komoře totiž může zajistit, že klidový systolický volum anebo srdeční výdej budou normální i u pacienta trpícího srdeční nedostatečností.
Hodnoty konečného diastolického objemu a ejekční frakce se nejčastěji rutinně stanovují echokardiograficky. Je možno použít i jiné přístupy (např. izotopová vyšetření). Srdeční výdej lze rovněž měřit metodami opírajícími se o Fickův princip (PiCCO, LiDCO).
Normální hodnoty:
- Normální hodnoty konečného komorového diastolického objemu jsou 110 – 120 ml (přesněji 70 ± 20 ml/m2).
- Normální ejekční frakce (poměr systolický objem/konečný diastolický objem) je 0,6 – 0,7 (přesněji 0,67 ± 0,08). Za dolní hranici normy se považuje ejekční frakce 0,5. Frakce rovná 0,45 – 0,5 anebo vyšší se označuje jako téměř normální.
Diagnostickou hodnotu uvedených vyšetření limituje skutečnost, že jejich výsledky jsou významně závislé na mimosrdečních faktorech. End-diastolický volum, ejekční frakce a srdeční výdej se mohou měnit jak při sníženém předtížení (např. pokles plnění srdce a srdečního výdeje při hypovolémii), tak při zvýšeném dotížení (např. zvýšené plnění srdce a pokles ejekční frakce při stenóze aorty anebo při hypertenzi), a to i při naprosto zdravém srdci.
Zátěžová vyšetření
Zátěžová vyšetření nejčastěji využívají farmakologickou zátěž, bicykl anebo pohyblivý chodník. Podobně jako při klidových vyšetřeních je jejich výhodou možnost spojit je s měřením spotřeby kyslíku, nebo s vyšetřením hemodynamických veličin katetrizací, elektrokardiografií, echokardiogragií anebo scintigrafií apod.
Intervenční zátěžová vyšetření
Normální stav:
- End-diastolický tlak v levé komoře v klidu je menší než 12 mm Hg;
- End-diastolický tlak v levé komoře se při námaze příliš nemění;
- Systolický objem při námaze roste, zejména při zátěži ve vzpřímené poloze;
- Srdeční výdej při námaze roste, a to nejméně o 500 ml krve/min na každých 100 ml O2/min přírůstku spotřeby kyslíku.
Patologický stav (selhávající komora):
- End-diastolický tlak v levé komoře při námaze roste nad 12 mm Hg;
- Systolický objem při námaze zůstává stejný, nebo dokonce klesá;
- Srdeční výdej při námaze roste příliš málo - o méně než o 500 ml krve/min na každých 100 ml O2/min přírůstku spotřeby kyslíku.
Tělesná spotřeba kyslíku při zátěži
Normální tělesná spotřeba kyslíku při vystupňované zátěži roste o více než 20 ml O2/min/kg tělesné hmotnosti. Hodnoty pod 10 ml O2/min/kg jsou výrazně patologické. Svědčí pro těžké postižení srdce s nepříznivou prognózou.
Spotřeba kyslíku v srdci
Při systolickém selhání s nízkým srdečním výdejem klesá podíl vnější práce vykonané myokardem, zatímco spotřeba kyslíku zůstává přibližně na stejné úrovni; poměr obou veličin, který charakterizuje výkonnost myokardu, se tedy snižuje.
Ukazatel spotřeby kyslíku – plocha systolický tlak-objem – roste při zvýšeném dotížení, a koreluje se spotřebou O2. Jak ukazuje obrázek 13, je za to především odpovědný nárůst vnitřní práce při vzrůstu dotížení, zatímco vnější práce sama může při vzrůstu dotížení klesat.

Obr. 13. Růst vnitřní práce při zvýšeném dotížení. (Podle Harrison´s Principles of Internal Medicine, 15th Ed. McGraw-Hill, New York, 2001.).
Laboratorní biochemická vyšetření
- Stanovení ukazatelů metabolismu kyslíku (arteriovenózní diference).
- Stanovení plazmatické koncentrace mozkového natriuretického peptidu (BNP). Plazmatická koncentrace BNP roste jak při systolickém, tak při diastolickém selhání. Podle některých studií BNP přímo koreluje se závažností diastolické dysfunkce levé komory.
Vyšetření diastolické funkce (vyšetření lusitropie)
Největší diagnostický přinos pro posouzení diastolické funkce komor mají:
- Echokardiografie kombinovaná s dopplerovnským vyšetřením
- Izotopová vyšetření.
Oba přístupy dovolují změřit rychlost průtoku krve atrioventrikulárními ústími a porovnat je s normou a také stanovit poměr rychlosti plnění komory v časné a v pozdní diastolické fázi.
Rychlost plnění komor ve zdravém srdci je největší na počátku systoly. Proto je poměr rychlosti v časné fázi k rychlosti v pozdní fázi větší než 1. Pokud vázne relaxace komory, roste podíl pozdní pasivní presystolické fáze diastoly a posiluje se význam atriálního stahu. Hodnoty poměru menší než 1 proto signalizují poruchu plnění. V pokročilých fázích městnání, kdy tlak před selhávající komorou významně roste, se však situace znovu obrací ve prospěch rané fáze („pseudo-normální“ nález). Potom má význam vyhodnotit jiné parametry, např.:
- Trvání izovolumové relaxační fáze (prodloužení je známkou diastolické dysfunkce);
- Rychlost systolického a diastolického toku krve v plicních žilách;
- Měření pohybu atriovenózního prstence. Je nejspolehlivější neinvazivní metodou hodnocení diastolického děje; vyžaduje zařízení pro tzv. tkáňové doplerovské zobrazení (TDI; Tissue Doppler Imaging).
Nejpřesnější údaje o diastolickém procesu lze získat pouze invazivně změřením plnicích tlaků a konstrukcí tlakově-objemové křivky (viz obrázek 13 nahoře).
Zpracovali: Jaroslav Veselý, Ústav patologické fyziologie LF UP, a Martin Hutyra, 1. interní klinika LF UP a FN v Olomouci


{kind=link}
{kind=link}