

Metabolismus bilirubinu
Normální koncentrace bilirubinu v plazmě dospělých je 5 – 17 µmol/l, z toho pouze 1 – 5 µmol/l tvoří konjugovaný bilirubin.
Hlavním zdrojem plazmatického bilirubinu je hem erytrocytů, které zanikají v orgánech, v nichž jsou nakupeny mononukleární makrofágy (retikuloendotelový systém). Cyklická struktura hemu je štěpena hemoxidázou a produkt štěpení, biliverdin, je redukován biliverdinreduktázou na bilirubin; vedlejším produktem štěpení hemu je oxid uhelnatý:
- Hem → CO + biliverdin → bilirubin
Asi 10 – 30 % bilirubinu pochází z jiných hemoproteinů, než je hemoglobin, anebo z mezistupňů neefektivní erytropoézy.
Bilirubin vstupuje do krevní plazmy jako nekonjugovaný. Je lipofilní, špatně rozpustný ve vodě. V plazmě koluje vázaný především na albumin. Nekonjugovaný bilirubin je transportován do jater, kde difunduje napříč povrchovými membránami krevního pólu jaterních buněk do cytoplazmy hepatocytů. Tam ho přebírá vazebný, transportní a katalytický protein glutathion-S-transferáza B (GST; dříve popisovaná jako ligandin). Usměrňuje ho k membránám endoplazmatického retikula, kde dochází k jeho detoxikaci konjugací.
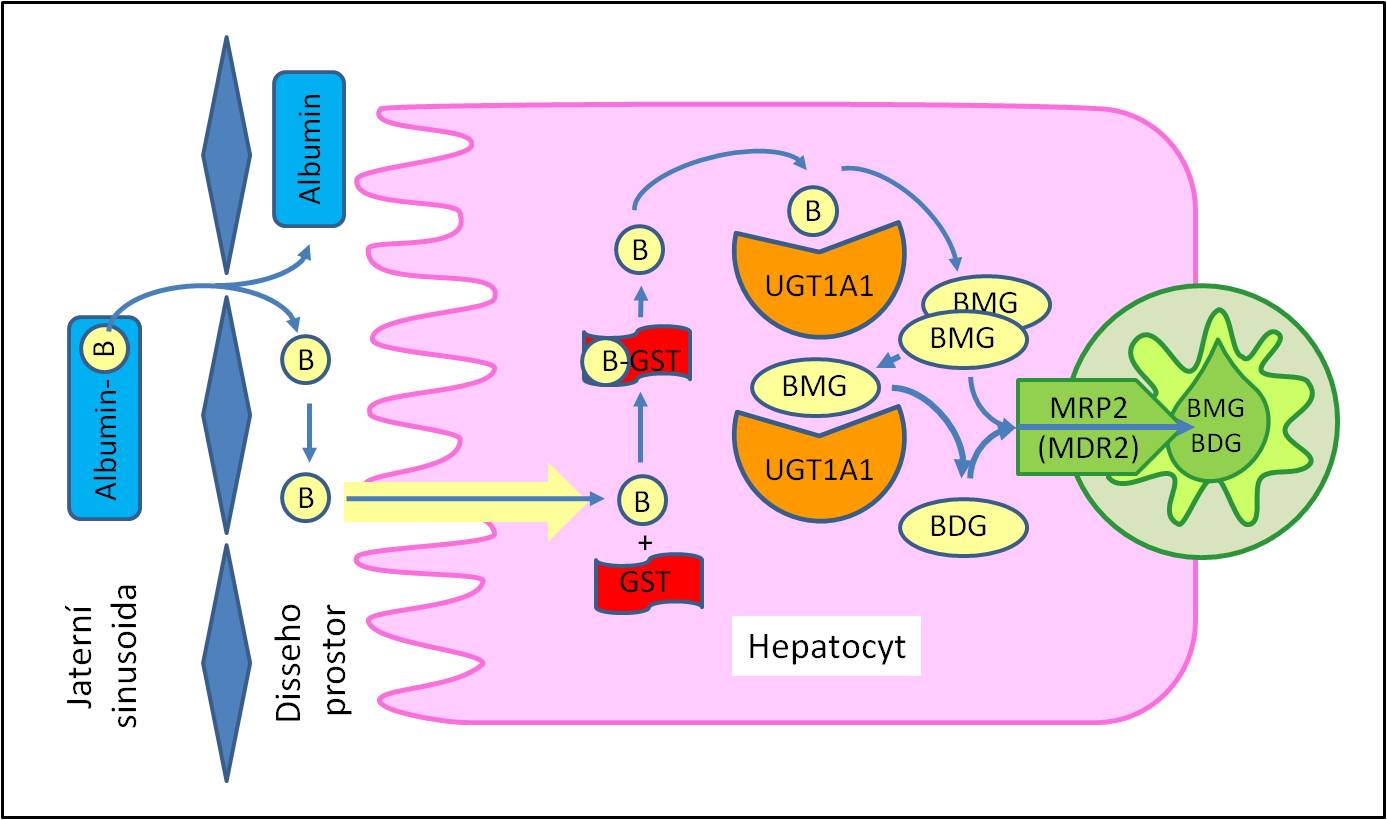
Obr. 1. Metabolismus bilirubinu. B, bilirubin; BDG, bilirubindiglukuronid; BMG, bilirubinmonoglukuronind; GST, glutathion-S-transferáza (ligandin); UGT; uridindifosfátglukuronyltransferáza.
Kapacita albuminu pro vazbu bilirubinu má svůj limit. Pokud je tento limit překročen, bilirubin se hromadí v lipofilních strukturách, zejména v nervové tkáni. Asi nejtěžší následky vznikají u novorozenců při masívní hemolýze spojené s inkompatibilitou Rh faktoru (jádrový ikterus). Nekonjugovaný bilirubin se nikdy nevyskytuje v moči, ani při vysoké hyperbilirubinémii. Normálně se nevyskytuje ani ve střevě, vyjma dvou případů:
- U novorozenců, u nichž teprve vyzrávají detoxikační pochody;
- Při těžké nekonjugované hyperbilirubinémii Crigler-Najjarova syndromu typu I.
K detoxikaci bilirubinu v jaterních buňkách dochází hlavně jeho konjugací s kyselinou glukuronovou za účasti UDP-glukuronyltransferáz (UGT). Asi 7 % konjugovaného bilirubinu je monoglukuronid, 90 % normálně je ve formě diglukuronidu. Konjugovaný bilirubin je aktivně vylučován biliárním pólem hepatocytů do žlučových kanálků. Aktivní transport zprostředkuje membránový transportér z nadrodiny ABC proteinů (ATP-binding casette proteins) známý jako MDR2 nebo MRP2 (multidrug resistence-associated protein 2). Konjugovaný bilirubin normálně tvoří méně než 1/5 celkového plazmatického bilirubinu. Transport do žlučových kanálků v játrech však je mimořádně citlivý k poškození, a pak dochází k přestupu primární žluči do krve. Konjugovaná hyperbilirubinémie tedy je abnormální jev. Ukazuje na poruchu transportu konjugovaného bilirubinu z hepatocytů do žlučových cest.
Bakterie přítomné v ileu a v tlustém střevě mění bilirubin na urobilinobgen a dále na urobilin. Urobilinogen podléhá enterohepatální cirkulaci. Ze střeva se vstřebává do krve, odkud se může znova vyloučit buď jaterní cestou do žluče, nebo ledvinami do moče. Urobilinogen proto je normální složkou moče. Ke změnám vylučování močí může dojít v následujících případech:
- Při patologickém postižení hepatocytů, kdy dochází ke snížení podílu urobilinogenu vylučovaného do žluči, a proto se zvyšuje podíl vylučovaný močí;
- Při hemolýze, kdy je vysoká nabídka plazmatického bilirubinu a následně i urobilinogenu, takže roste i vylučování urobilinogenu ledvinami.
- Cholestáza naopak vede ke sníženému obsahu bilirubinu a urobilinogenu ve střevě, takže klesá i enterohepatální oběh urobilinogenu a jeho a vylučování do moče;
- Podobný účinek má i potlačení střevní flóry antibiotiky, které rovněž snižuje nabídku urobilinogenu a jeho vylučování močí.
Nekonjugovaná hyperbilirubinémie
Nekonjugovaná hyperbilirubinémie může vznikat buď z důvodu zvýšené tvorby bilirubinu, nebo ztížené detoxikace bilirubinu. S první skupinou poruch se setkáváme při hemolýze a při poruchách tvorby erytrocytů. Druhá skupina zahrnuje následky poruch transportu nekonjugovaného bilirubinu na krevním pólu hepatocytů a z poruch konjugace. Nekonjugovaná hyperbilirubinémie je spojena s vysokou náloží konjugovaného bilirubinu směřujícího do střeva a následně i urobilinogenu podléhajícího enterohepatálnímu oběhu. Výsledkem jsou zvýšená množství urobilinogenu v moči.
Nekonjugované hyperbilirubinémie z vysoké nabídky bilirubinu
Hemolýza
Krevní dřeň může v případě potřeby 8krát dlouhodobě zvýšit produkci erytrocytů. To může mít za následek mírnou nekonjugovanou hyperbilirubinémii; pokud jsou koncentrace nekonjugovaného bilirubinu při hemolýze abnormálně vysoké, ukazují na souběžnou poruchu jaterních funkcí. Je vhodné pamatovat, že velké množství nekonjugovaného bilirubinu se také může uvolňovat při vstřebávání velkých hematomů.
Neefektivní erytropoéza
Neefektivní erytropoéza provází velkou skupinu anémií vznikajících z poruch maturace; je spojena s talasémií, porfyrií, otravami těžkými kovy aj. Tvorba bilirubinu z nehemových zdrojů může dosáhnout až 70 % nekonjugovaného bilirubinu.
Nekonjugované hyperbilirubinémi z poruch transportu nekonjugovaného bilirubinu na krevním pólu hepatocytů a z poruch konjugace
Některé přirozené metabolity anebo léčiva mohou soutěžit s bilirubinem o transport krevním pólem hepatocytů anebo o průchod konjugačním aparátem. Jejich přítomnost může způsobit nekonjugovanou hyperbilirubinémii. Lze uvést následující příklady:
- Glutation-S-transferáza B (ligandin) může vzhledem ke své malé vazebné specifičnosti vázat četné endogenní i exogenní ligandy, které mohou soutěžit s bilirubinem, bránit jeho vazbě, a způsobit nekonjugovanou hyperbilirubinémii;
- Mateřské mléko obsahuje mastné kyseliny a progesteronový steroid 3α,20β-pregnandiol. Obojí může inhibovat konjugaci (žloutenka z mateřského mléka, breast milk jaundice).
Fyziologická žloutenka novorozenců se vyvíjí během 2. – 5. dne po porodu a obvykle odeznívá do dvou týdnů. Jde o souběh několika faktorů:
- Vysoká nabídka nekonjugovaného bilirubinu pochází z výměny krvinek s fetálním hemoglobinem za krvinky obsahující hemoglobin dospělých.
- Jaterní konjugační aparát novorozenců není vyzrálý, koncentrace UDP-glukuronyltransferáz je nízká, takže do střeva přechází i nekonjugovaný bilirubin.
- V gastrointestinálním traktu novorozených chybí intestinální mikroflóra, která by přeměnila bilirubin na urobilinogen. Podíl bilirubinu cirkulujícího mezi střevem a játry tak je mnohem vyšší než u dospělých.
- Biliární pól hepatocytů a funkce žlučových kanálků v játrech novorozenců nejsou plně rozvinuty, takže se může objevit i přechodná konjugovaná hyperbilirubinémie.
Masivní žloutenka novorozenců se vyvíjí při hemolýze způsobené inkompatibilitou Rh-faktoru. Může způsobit bilirubinovou encefalopatii (jádrový ikterus). Léčebným řešením je prevence, (intrauterinní) krevní transfúze a fototerapie.
Nekonjugované bilirubinémie z vrozených defektů konjugace
Gilbertův syndrom
Jde o relativně častou poruchu. Její výskyt v populaci je asi 8 %, přičemž převažují muži. Hyperbilirubinémie bývá mírná a zvyšuje se po zátěži, kterou může být námaha, hladovění, alkohol anebo jiná nemoc. Příčinou je výrazný deficit UDP-glukuronyltransferáz, jejichž obsah v jaterních buňkách dosahuje pouze 10 – 35 % normálního množství. Postižené osoby špatně tolerují řadu léčiv, která snadno mohou dosáhnout toxických koncentrací.

Obr. 2. Přehled vybraných forem nekonjugované hyperbilirubinémie. Přehled vybraných forem konjugované hyperbilirubinémie. B, bilirubin; BDG, bilirubindiglukuronid; BMG, bilirubin monoglukuronid; GST, glutathion-S-transferáza; MRP2 (MDR2), multidrug resistance-associated protein 2; UGT, uridindifosfátglukuronyltransferáza.
Crigler-Najjarův syndrom typu II
Deficit UDP-glukuronytransferáz a jemu odpovídající hyperbilirubinémie jsou ještě více vyjádřený než v předchozím případě (v hepatocytech je pouze 10 % jnormálního množství UDP-glukuronyltransferáz. Konjugací vznikají hlavně monoglukuronidy. Jejich stanovení je základem diagnózy.
Crigler-Najjarův syndrom typu I
Jde o velmi vážnou formu nekonjugované hyperbilirubinémie, která se objevuje už záhy po narození, aniž je zvýšená hemolýza. Do žluče a tenkého střeva se přímo vylučuje nekonjugovaný bilirubin. V moči bilirubin není. Urobilinogen ve stolici anebo v moči se vyskytuje jen ve stopách. Dochází k těžkému postižení nervového systému. Ulevit může fototerapie nebo transplantace jater.
Konjugovaná hyperbilirubinémie
Níže jsou zmíněny pouze vybrané konjugované hyperbilirubinémie. Kromě primární biliární cirhózy jsou způsobeny vrozenými defekty.
Rotorův syndrom
Při tomto onemocnění je mírná konjugovaná hyperbilirubinémie vyvolána dědičnou změnou transportního proteinu MRP2. Porucha není vážná. Není ani vážněji postiženo vylučování jiných sloučenin, závislých na MRP2, takže je možno zobrazit rtg-kontrastní látku procházející biliárním kanalikulárním systémem.
Dubin-Johnsonův syndrom
Je rovněž vyvolán mutací MRP2, porucha se však manifestuje nápadněji než předchozí onemocnění. Je přítomna mírná konjugovaná hyperbilirubinémie s bilirubinurií. Exkrece endogenních i exogenních aniontů včetně léků a rtg-kontrastních sloučenin je postižena hůře než u Rotorova syndromu, takže je zapotřebí pamatovat na zvýšenou toxičnost léčiv. Pro zobrazení žlučových cest nelze použít rtg-kontrastní látky. Charakteristická je abnormální přítomnost pigmentu v jaterních buňkách.
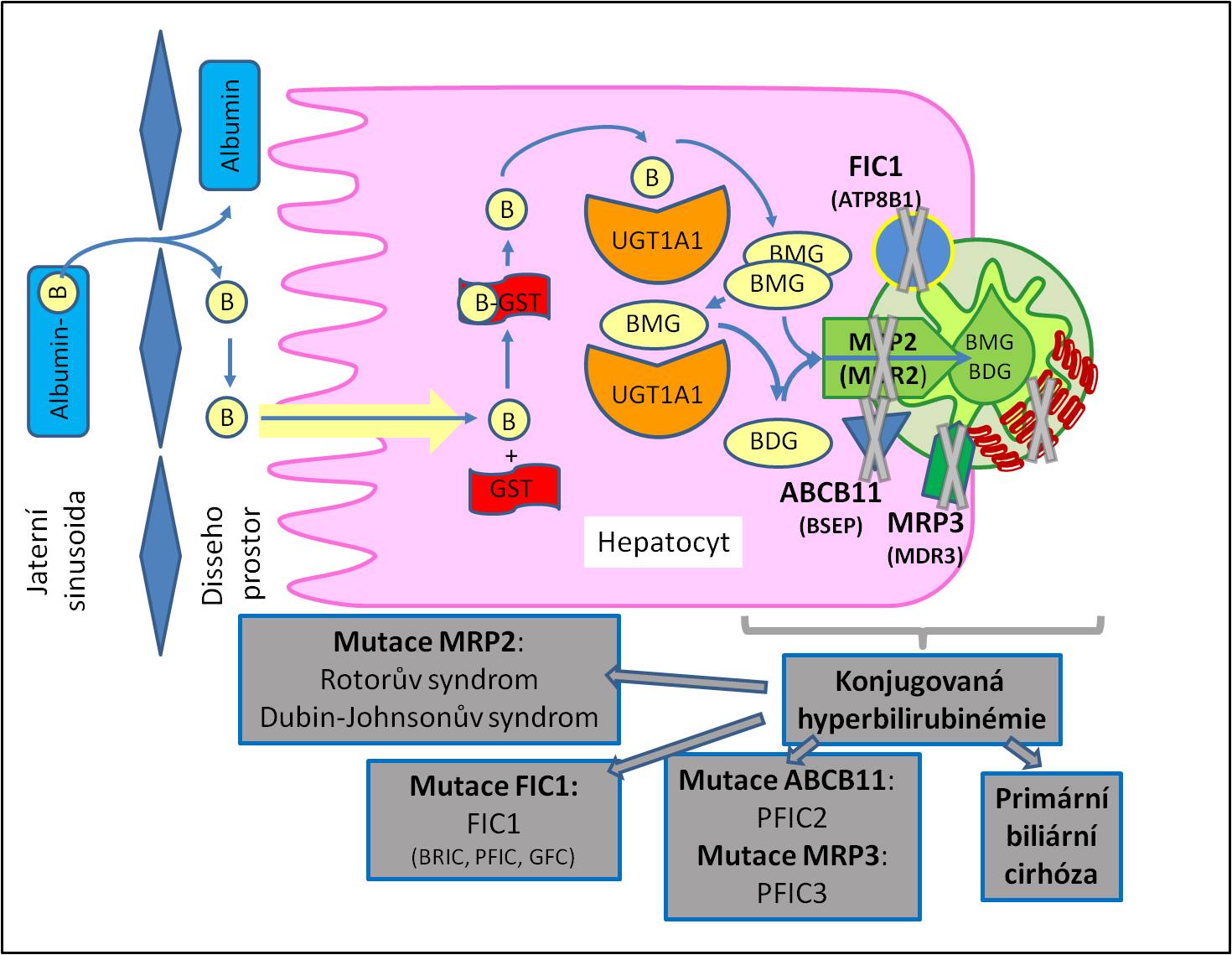
Obr. 3. Přehled vybraných forem konjugované hyperbilirubinémie. B, bilirubin; BDG, bilirubindiglukuronid; BMG, bilirubin monoglukuronid; FIC1 (ATP8B1), protein familiární intrahepatické cholestázy – ATP-dependentní aminofosfolipidová translokáza – familiární intrahepatická cholestáza 1; ABCB11 (BSEP), bile salt export protein; GST, glutathion-S-transferáza; MRP2 (MDR2), multidrug resistance-associated protein 2; MRP3 (MDR3), multidrug resistance-associated protein 3; PFIC2, progresivní familiární intrahepatická cholestáza 2; PFIC3, progresívní familiární intrahepatická cholestáza 3; UGT, uridindifosfátglukuronyltransferáza.
Familiální intrahepatická cholestáza typu 1
Sdružuje tři formy. První je benigní návratná (rekurentní) intrahepatická cholestáza (BRIC), druhá je progresivní familiární intrahepatická cholestáza (PFIC), třetí Grónská familiální cholestáza (GFC). Zahrnutí všech tří uvedených onemocnění pod jedním označením je podloženo geneticky. Všechny jsou způsobeny defekty membránového transportéru rodiny P-typu ATPáz, který katalyzuje přemístění aminofosfolipidů z vnější strany plazmatické membrány na stranu cytoplazmatickou. Tato ATP-dependentní aminofosfolipidová translokáza nese označení ATP8B1 neboli FIC1 (podle familial intrahepatic cholestasis). Diferenciálně diagnosticky jde o cholestázy (hyperbilirubinémie) s nezvýšenou plazmatickou koncentrací γ-glutamyltransferázy. Alaninaminotransferáza, alkalická fosfatáza a bilirubin bývají zvýšené, přinejmenším na počátku ataky.
Benigní návratná (rekurentní) intrahepatická cholestáza (BRIC) je charakteristická opakujícími se cholestatickými příhodami, které se manifestují svěděním a žloutenkou. V případě PFIC a GFIC mají poruchy progresívní průběh a způsobují těžké chronické poškození jater s průvodní malabsorpcí anebo zpomalením růstu v dětství.
Progresivní familiální intrahepatická cholestáza typu 2 (PFIC2)
Je způsobena mutací transportního proteinu žlučových solí (ABCB11 neboli BSEP, bile salt export protein). Gama-glutamyltransferáza není zvýšena. Porucha má progresívní průběh a způsobují těžké chronické poškození jater s průvodní malabsorpcí anebo zpomalením růstu v dětství.
Progresivní familiální intrahepatická cholestáza typu 3 (PFIC3)
Je způsobena mutací genu MRP3 neboli MDR3 (multidrug resistence-associated protein 3). Žluč postrádá fosfatidylcholin a je mimořádně agresivní už ve žlučových kanálcích. Gama-glutamyltransferáza je výrazně zvýšena. Porucha má progresívní průběh a způsobuje těžké chronické poškození jater s průvodní malabsorpcí anebo zpomalením růstu v dětství. Je vysoké riziko tvorby žlučových kamenů.
Primární biliární cirhóza
Při tomto závažném onemocnění se žlučové cesty stávají objektem nehnisavé autoimunitní reakce. Primárně je napadán epitel lemující jemné žlučovody, takže morfologické změny začínají v portálních a pak periportálních polích. Až v 90 % jsou postiženy ženy. Progrese do cirhózy naštěstí není tak častá, jak se dříve myslelo. Mnozí autoři proto soudí, že název nevystihuje povahu onemocnění.
Zpracoval: Jaroslav Veselý, Ústav patologické fyziologie LF UP, a Jiří Ehrmann, 1. Interní klinika LF UP a FN v Olomouci

{kind=link}
{kind=link}