

autor: MUDr.Ondřej Veselý
pracoviště: Ústav patologické fyziologie LF UP Olomouc, Dětská endokrinologická ambulance Svitavské nemocnice a.s.
Glukokortikoidy a základní přehled jejich fyziologických účinků
Glukokortikoidy jsou členy široké skupiny steroidních hormonů, mezi jejichž základní společné znaky patří:
- Biochemický původ – prekurzorem pro tvorbu steroidů je cholesterol. Endokrinní buňky tvořící steroidní hormony jsou proto v bohaté míře vybaveny receptory pro LDL částice, pomocí kterých získávají potřebný cholesterol.
- Mechanismus působení – Steroidní hormony jsou lipofilní a procházejí skrze buněčnou membránu, aniž by k tomu potřebovaly membránové transportéry. V cytoplazmě nacházejí své receptory, se kterými se spojují. Vzniklý komplex steroid-steroidní receptor se přesouvá do buněčného jádra, kde se váže na příslušnou hormon responzivní oblast DNA. Cílová oblast je součástí promotoru steroidem regulovaného genu, po navázání výše uvedeného komplexu dochází ke stimulaci nebo inhibici transkripce cílového genu. Změna transkripce vyžaduje čas a tak není divu, že účinky steroidních hormonů se dostavují řádově v hodinách, ale současně je jejich působení dlouhodobé. Nicméně steroidy jsou schopné vyvolat v organismus i odpovědi v během vteřin a minut, tyto negenomové účinky jsou zprostředkované přes membránové receptory za spoluúčasti G-proteinů a přes změnu aktivity intracelulárních signalizačních kaskád prostřednictvím různých kináz a fosfatáz.
Hlavním lidským glukokortikoidem je kortisol (jiným názvem hydrokortison), který je vytvářen v zona reticularis kůry nadledvin. Kortisol je hormon se širokou paletou účinku:
- Účinky metabolické – zasahují do přeměny všech základních živin, tedy nejen cukrů, ale i tuků a bílkovin. Tyto účinky probereme podrobněji v další kapitole, ve zkratce lze konstatovat, že kortisol vede ke katabolismu, k aktivaci energetických zdrojů a zvýšení hladiny krevního cukru. Metabolické účinky kortisolu patří mezi jeho účinky genomové.
- Účinky na imunitní systém – zde kortisol zasahuje na mnoha místech, výsledný vliv lze shrnout jako účinek protizánětlivý a imunosupresivní. Tyto účinky patří svou podstatou mezi negenomové, takže kortikoidy lze užít nejen k léčbě chronických zánětlivých chorob, ale i ve vybraných akutních stavech jako jsou bouřlivější alergické reakce včetně anafylaxe a těžšího záchvatu bronchiálního astmatu. Dále třeba využití efektu antiedematozního při akutní laryngitidě u dětí nebo u míšního šoku.
- Jiné účinky – Sem můžeme zařadit účinky kortisolu na CNS, které začínají už prenatálně, kdy se kortisol podílí na vývoji mozku, tak postnatálně svým účinkem na tvorbu paměťových stop.
Metabolické účinky kortisolu
Kortisol je hormonem stresu a jednou ze základních charakteristik stresové odpovědi je mobilizace energetických zdrojů pro boj se stresorem, což v 1. fázi stresové odpovědi zajišťují katecholaminy a ve 2. fázi právě glukokortikoidy. Mobilizované energetické zdroje pak směřují do orgánů potřebných pro přežití tedy do mozku (glukosa), myokardu a pracujících svalů (volné mastné kyseliny, méně glukosa).

Srovnání metabolických účinků inzulinu a kortisolu
Jak již bylo řečeno výše, je účinek kortisolu charakterizován zvýšeným katabolismem základních živin. Kortisol mění metabolismus mnoha tkání, ale klíčové jsou tři a to játra, svaly a tuková tkáň, podobně jako je tomu v případě inzulinu a proto metabolické účinky kortisolu probereme „orgánově“.
- Játra. Hlavním hepatálním a současně i hlavním metabolickým účinkem kortisolu je stimulace glukoneogeneze, tedy novotvorby glukosy z nesacahridových substrátů, jako jsou aminokyseliny, laktát/pyruvát, glycerol. Kortizol zvyšuje tvorbu a výdej glukosy z jater stimulací exprese klíčových enzymů glukoneogeneze, kterými jsou: (a) fosfoenolpyruvát-karboxykináza (PEPCK), která katalyzuje přeměnu oxalacetátu na fosfoenolpyruvát a (b) glukosa-6-fosfatáza, která hydrolyzuje fosfát z molekuly glukosa-6-fosfátu (Glu-6-P) , tím se uvolní glukosa (Glu), která je schopná (na rozdíl od Glu-6-P) po koncentračním gradientu opustit jaterní buňku, což zvyšuje glykemii. Pro glukoneogenezi je potřeba zajistit výše uvedené substráty a s tím souvisí další účinek kortisolu v játrech a to stimulace syntézy mastných kyselin (stimulace exprese syntázy MK a acetyl-CoA karboxylázy) a současně snížení jejich spotřeby inhibicí β-oxidace mastných kyselin. Jako vedlejší, ale žádoucí efekt syntézy MK je spotřeba acetyl-CoA, a tím snížení aktivity Krebsova cyklu, což zvyšuje dostupnost oxalacetátu pro glukoneogenezi.
- Kosterní svalovina představuje hlavní zásobárnu bílkovin našeho organismu a tudíž i potencionální zdroj glukoplastických aminokyselin pro glukoneogenezi. Kortizol jako katabolický hormon zvyšuje proteolýzu svalových bílkovin a současně inhibuje proteosyntézu. Zvýšená degradace proteinů vlivem kortisolu je dána především aktivací ubiquitin-proteasomové cesty. Kde ubiquitin je drobný protein, který funguje jako „cejch smrti“, bílkovina jím „označená“ je rozložena uvnitř proteasomu, bílkovinného komplexu s proteolytickou aktivitou. Blokáda proteosyntézy kortizolem souvisí s jeho inhibičním účinkem na kaskádu začínající aktivací receptorů pro insulin a pro IGF-1. Inzulin a IGF-1 jsou naopak proteoanabolické působky podporující nárůst svalové hmoty. Blokáda jejich účinku kortizolem na receptorové (snížení tyrosin-kinázové aktivity intracelulární domény receptorů pro insulin a IGF-1) a postreceptorové úrovni (inhibice fosforylačních enzymů insulin/IGF-1 signální cesty vedoucí k aktivaci nebo deaktivaci mnoha transkripčních faktorů) vede k rozkladu svalových bílkovin s uvolněním aminokyselin, které jsou vychytávány v játrech a použity pro glukoneogenezi. Další účinek kortisolu ve svalech je zacílen přímo na metabolismus cukrů. Jelikož mozek dokáže jako zdroj energie využívat jen glukosu (a ketolátky), a nikoliv mastné kyseliny, tak kortisol snižuje vychytávání glukosy svaly a to snížením počtu glukosových transportérů (GLUT4) v membráně svalových buněk a také snižuje tvorbu glykogenu inhibicí glykogensyntázy, čímž snižuje spotřebu glukosy ve svalech ve prospěch CNS.
- Tuková tkáň má dvě metabolicky odlišně se chovající kompartmenty a to tuk podkožní a tuk viscerální. Účinek kortisolu je v obou oddílech tělesného tuku odlišný. V periferní tukové tkáni (tuk podkožní) kortisol zvyšuje lipolýzu zvýšením aktivity hormon-senzitivní lipázy, která katalyzuje rozklad uložených triglyceridů, a současně kortisol blokuje lipogenezi (a) inhibicí aktivity lipoproteinoví lipázy, která sedíce na endotelu rozkládá triglyceridy (TAG) lipoproteinových částic jako jsou chylomikry či VLDL a uvolněné mastné kyseliny jsou pak uvnitř adipocytu použity na syntézu TAG a tyto uloženy do zásoby, (b) inhibicí PEPCK, která se mimo výše popsanou úloho v glukoneogenezi (játra, ledviny) také účastní procesu glyceroneogeneze (tuková tkáň) za vzniku glycerol-trifosfátu jenž se esterickou vazbou spojuje s mastnými kyselinami za vzniku mono-,di-,triglyceridů. V centrální (viscerální) tukové tkáni je účinek kortisolu zcela opačný, zde podporuje lipogenezi stimulací diferenciace pre-adipocytů ve zralé adipocyty a v nich zvyšuje syntézu triglyceridů.
Regulace působení kortisolu
Účinek kortisolu závisí na několika faktorech:

Osa hypothalamus-hypofýza-kůra nadledvin
- Kortisolemie – tedy plazmatická koncentrace kortisolu. Ta je udržována pomocí osy hypothalamus – hypofýza – adrenocortex (HHA) mechanismem negativní zpětné vazby. Snížení hladiny kortisolu vede k tvorbě a vyplavení kortikoliberinu (CRH) z hypothalamu přesněji z malobuněčné části ncl. paraventricularis. CRH je hormon peptidické povahy (41 aminokyselin), který v kortikotrofních buňkách adenohypofýzi nesoucích na svém povrchu receptory pro CRH (CRH-R1) zvyšuje tvorbu adrenokortikotropního hormonu (ACTH) neboli kortikotropinu. ACTH je rovněž peptidický hormon (39 AMK) a vzniká z větší prekurzorové molekuly propiomelanokortinu (POMC). ACTH se pak krevním oběhem dostává do kůry nadledvin, kde zvyšuje tvorbu kortisolu, nadledvinových androgenů (androstendion a dehydroepiandrosteron) a v menší míře i aldosteronu (hlavním stimulem pro aldosteron jsou ioty K+ a angiotenzin II). Účinek ACTH na buňky kůry nadledvin je zprostředkován přes melanokortinové receptory (MCR). Jelikož steroidní hormony nejsou syntetizovány do zásoby, tak se hladina kortisolu po stimulaci kortikotropinem zvyšuje s latencí několika desítek minut až hodin.
- Cirkadiální rytmus – hladina kortisolu kolísá v průběhu dne, kdy nejvyšší bývá okolo 8. hodiny ranní a nejnižší okolo půlnoci. Kromě ranního vrcholu má kortisolemie ještě další 2 a to okolo poledne a dále v podvečer okolo 16 – 17 h, tyto jsou co do absolutní výše nižší než ranní maximum. Cirkadální rytmus je určován především vnitřními hodinami organismu, jejichž centrem ncl. suprachiasmaticus. Dále se na cirkadiálním rytmu podílí potřeba udržet stabilní glykemii v době mezi jídly, proto je nejvyšší hladina kortisolu ráno, protože v noci kdy hladovíme je glykemie udržována nejprve glykogenolýzou v játrech a po vyčerpání zásob glykogenu játra přepínají na glukoneogenezi, jejíž klíčové enzymy kontroluje-stimuluje právě kortisol. Lidově řečeno, abychom byli schopni se ráno postavit na zadní a dojít k ledničce, tak potřebujeme ranní peak kortisolu.
- Hladina kortikosteroidy vázajícího globulinu (CBG). Jak mnohé hormony kolují v krvi ve dvou základních frakcích. Frakci vázané na plazmatické bílkoviny: (a) obvykle nespecificky a se slabší afinitou na albumin, (b) specificky a silně na příslušný globulin (TBG pro hormony štítné žlázy, SHBG pro hormony pohlavní anebo třeba právě CBG pro kortisol). Frakci volné, která ale je biologicky účinná.V případě kortisolu je na CBG vázáno 75 – 80 % kortisolu, na albumin asi 15 % a volná frakce tvoří 5 – 10 %. Hladina CBG tedy reguluje dostupnost kortisolu buňkám a tím i jeho působení. Hladinu CBG stimuluje samotný kortisol nebo třeba estrogeny.
- Intracelulární hladina kortisolu v cílových buňkách. Nezávisle na plazmatické koncentraci kortizolu je regulována biologická aktivita kortisolu na úrovni cílových buněk podle jejich potřeb buď přeměnou biologicky aktivního kortisolu na biologicky neaktivní kortison nebo naopak konverzí neaktivního kortisonu na aktivní kortison. Tyto vzájemné přeměny katalyzuje enzym 11-β-hydroxysteroiddehydrogenáza (11-β-HSD).
Fyziologická úloha 11-β-HSD
11-β-HSD představuje klíč k tkáňově specifické postsekreční regulaci účinnosti kortisolu. Tím že je schopná konvertovat vzájemně mezi sebou aktivní kortisol a neaktivní kortison, mění dostupnost kortisolu pro glukokortikoidní receptory uvnitř buněk nezávisle na hladině kortisolu v krvi. Enzym se vyskytuje v lidském organismu ve dvou izoformách:
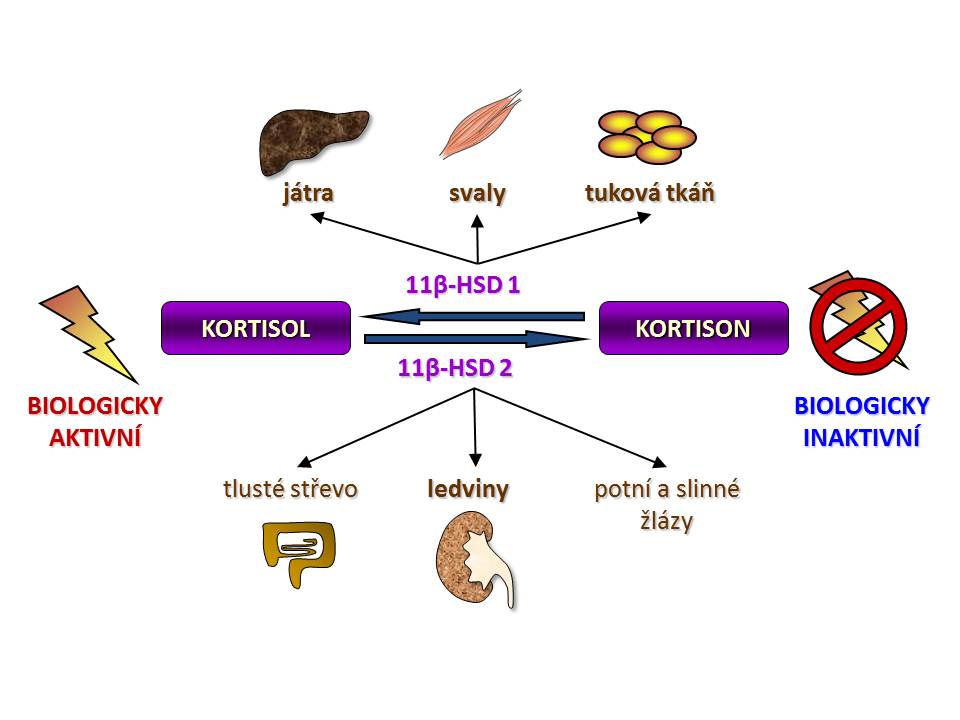
Úloha izoenzymů 11-beta-HSD a klíčové orgány jejich exprese
- 11-β-hydroxysteroiddehydrogenáza typ I (11-β-HSD1) je kódována genem na dlouhém raménku 1. chromozomu a v pokusech in vitro vykazuje jak reduktázovou aktivitu, tak aktivitu dehydrogenázovou, tzn., že je schopna obousměrné konverze kortison Û kortisol. Nicméně in vivo funguje hlavně jako reduktáza tzn. konvertuje neaktivní kortison na aktivní kortisol. Převaha reduktázové aktivity v organismu je dána nabídkou redukované formy NADPH, který funguje jako kofaktor reakce kortison Þ kortisol. Dodávku NADPH v endoplazmatickém retikulu, kde je 11-β-HSD lokalizována, zajišťuje „spolupracující“ enzym hexosa-6-fosfát dehydrogenáza (H6PDH), která oxiduje Glu-6-P na 6-fosfoglukonát (viz schéma). H6PDH je jinak součástí pentosového cyklu. 11-β-HSD1 je vysoce exprimována v řadě tkání především v játrech, tukové tkáni, kosterní svalovině, hypofýze, ale také v mozku, plicích, gonádách, hladké svalovině cév a dalších. Závěrem můžeme shrnout, že úkolem 11-β-HSD1 je intracelulární „recyklace“ kortisolu neboli zajištění dodávky kortisolu glukokortikoidním receptorům bez potřeby zvýšení sekrece a plazmatické koncentrace kortisolu.
- 11-β-hydroxysteroiddehydrogeníza typ II (1-β-HSD2) je kódována genem na dlouhém raménku 16. chromozomu a vykazuje striktně pouze dehydrogenázovou aktivitu, tzn., konvertuje aktivní kortisol na neaktivní kortison. Jejím kofaktorem je NAD+. 11-β-HSD2 se vyskytuje v aldosteron-dependentních tkáních, na prvním místě v ledvinách, přesněji řečeno ve stočené části distálních tubulů a ve sběracích kanálcích, dále se 11-β-HSD2 vyskytuje v tlustém střevě, potních a slinných žlázách. Proč právě v těchto orgánech? Protože smyslem existence 11-β-HSD2 je ochrana mineralokortikoidních receptorů před účinkem kortisolu, který je sice slabý mineralokortikoid, o 3 řády slabší než aldosteron, ale jeho koncentrace v plazmě je o asi 3 řády vyšší než koncentrace aldosteronu. Kortisol je tedy schopen se vázat na mineralokortikoidní receptory a pokud by 11-β-HSD2 nefungovala, docházelo by i za fyziologických plazmatických koncentrací kortisolu k aktivaci mineralokortikoidních receptorů ve výše uvedených tkáních, což by hlavně cestou ledvin vedlo k nežádoucí retenci sodíku s následným rozvojem hypertenze (viz níže pseudohyperaldosteronismy). 11-β HSD2 byla nalezena i v placentě (ochrana plodu před účinkem mateřského kortisolu?) nevbo v některých maligních nádorech (následek onkogenní transformace?).
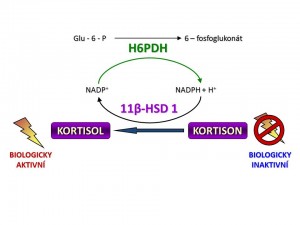
Spolupráce 11-beta-HSD s hexosa-6-fosfát-dehydrogenázou

Obezita jako epidemie 3. tisíciletí
Nárůst výskytu obezity dosáhl v rozvinutých zemích takřka úrovně epidemie a bude nepochybně jedním z největších zdravotnických problémů 21. století. V České republice trpí obezitou přes 20 % mužů a přes 30 % žen, pokud ale vezmeme v úvahu i předstupeň obezity tedy nadváhu pak vyšší než přiměřenou tělesnou hmotností je zatíženo okolo 70 % obyvatel ČR. Obezita zatěžuje naši společnost zvýšenou morbiditou a mortalitou postižených, což s sebou přináší obrovské finanční náklady spojené s léčbou obezity a jejich komplikací a finanční újmou společnosti při úmrtí člověka v produktivním věku. Hlavním problémem není samotná obezita, ale její komplikace a to především komplikace metabolické tj. nárůst výskytu cukrovky 2. typu a komplikace kardiovaskulární jako je hypertenze, ischemická choroba srdeční a cévní mozkové příhody. Tyto komplikace se navzájem potencují (cukrovka zvyšuje riziko vzniku infarktu myokardu, hypertenze zvyšuje riziko cévní mozkové příhody atd.). Není proto divu, že vědecký výzkum obezity a chorob s ní spojených je celosvětově velmi intenzivní. Výzkum se zaměřuje nejen na léčbu tohoto „balíku“ chorob s obezitou související, ale také je snahou mnoha studií pochopit etiopatogenezi a vzájemné souvislosti, protože jedině tak lze nalézt nové terapeutické přístupy.
Co mají společného obezita, cukrovka 2. typu, metabolický a Cushingův syndrom ?
80 % pacientů s cukrovkou 2. typu je obézních, 20 – 30 % obézních má cukrovku 2. typu. Mezi diagnostická kriteria metabolického syndromu patří přítomnost abdominální obezity + alespoň 2 další příznaky ze 4 možných (hypertenze, hyperglykemie/porucha glukosou tolerance, hypertriglyceridemie, nízký HDL cholesterol). Pacienti s Cushingovou chorobou mají typicky redistribuci tělesného tuku s redukcí podkožní tukové tkáně a nárůstem viscerálního tuku, trpí hypertenzí a může u nich dojít k vývoji steroidního diabetu.
Společným jmenovatelem všech jmenovaných chorobných stavů je akumulace centrálního-viscerálního tuku a základní endokrinní charakteristikou provázející nahromadění viscerálního tuku je inzulinorezistence, tedy snížená biologická odpověď tkání (kromě tukové tkáně také jater a kosterní svaloviny) vůči inzulinu.
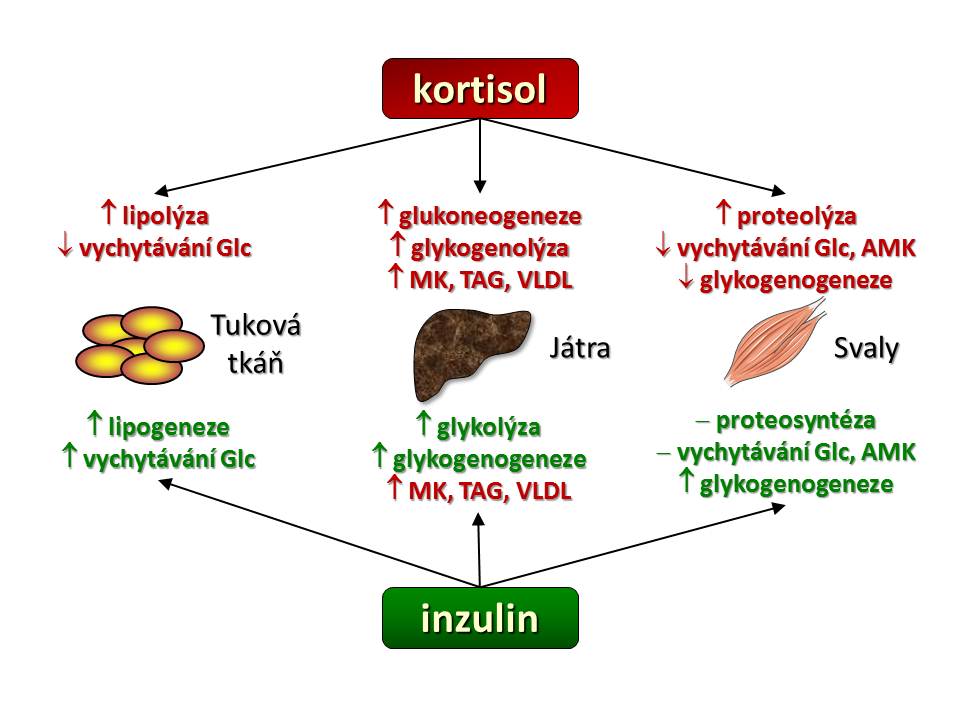
Srovnání metabolických účinků kortisolu a inzulínu
Pokud srovnáme účinky inzulinu a kortisolu (viz schéma), pak je zcela zjevné, že tyto dva hormony tvoří jakési protipóly, rub a líc v regulaci metabolismu základních živin. Zatímco inzulin je hormonem anabolickým podporujícím ukládání energie do zásob, tvorbu bílkovin, snižujícím glykemii, tak kortisol je hormonem katabolickým mobilizujícím energetické rezervy, zvyšujícím glykémii, hormonem vedoucím k odbourávání bílkovin a úbytku podkožního tuku. Ve vztahu k účinkům inzulinu můžeme kostatovat, že kortisol zvyšuje inzulinorezistenci a to nejen svým protichůdným účinkem na metabolismus cukrů, tuků a bílkovin, ale také svým přímým účinkem na beta buňky ostrůvků slinivky břišní, kde (a) snižuje sekreci inzulinu beta-buňkami, (b) zvyšuje apoptózu beta buněk a v neposlední řadě inhibičním účinkem na inzulinový signál v insulin-dependentních tkáních, kde (a) snižuje expresi IRS1 (insulin receptor substrate) proteinu, (b) zvyšuje degradaci IRS1, (c) zvyšuje fosforylaci IRS1 a inhibuje vedení inzulinového signálu v buňce.
V případě Cushingova syndromu je zvýšená hladina glukokortikoidů v krvi a tkáních příčinou inzulinorezistence a výsledného klinického obrazu. V případě prosté obezity, DM 2. typu a metabolického syndromu jsou příčiny inzulinorezistence komplexnější, nicméně dysregulace v sekreci a především změny metabolismu a aktivity glukokortikoidů v periferních tkáních byly u těchto onemocnění prokázány a zaujímají významné místo v jejich patogenezi.
11-β-HSD, kortizol a obezita
I prostá obezita je provázena změnami v sekreci kortisolu. O jaké změny se jedná? Celková sekrece kortisolu je zvýšená, což svědčí pro zvýšenou aktivitu HHA osy, ale samotné absolutní hladiny plazmatické kortisolemie obvykle nevybočují z širší normy, což svědčí pro zvýšený obrat kortisolu v periferních tkáních. Je porušen cirkadiální rytmus sekrece kortisolu, ranní maximum bývá nižší, podvečerní naopak vyšší. Na druhé straně také u obézních někdy vídáme ranní peak na horní hranici norem či dokonce lehce nad ní (norma závisí na laboratoři, obvykle se pohybuje mezi 220 – 690 nmol/l). Možným vysvětlením je, že obézní mají vyšší energetický výdej, což během nočního hladovění rychleji vyčerpá zásobu jaterního glykogenu a tudíž je potřeba nad ránem k udržení glykemie více „nažhavit“ glukoneogenezi, k čemuž je potřeba vyšší hladiny kortisolu. Dále je popisována zvýšená rezistence hypothalamu a hypofýzy na negativní zpětnou vazbu hladiny kortisolu.

Kritéria metabolického syndromu dle mezinárodní diabetologické federace (IDF)
Obezita je provázena kromě změn v sekreci i změnami v aktivitě glukokortikoidů v periferních tkáních a klíčem k tomuto je díky své funkci 11-β-HSD. Změna exprese a aktivity 11-β-HSD1 v metabolicky klíčových tkáních je proto zvažována jako jeden z možných a významných etiopatogenetických faktorů v rozvoji obezity a metabolického syndromu. Jaké změny v 11-β-HSD1 byly prokázány? Prvním krokem k tomuto poznání byly studie na animálních modelech. Transgenní myši s vystupňovanou expresí 11-β-HSD1 svým fenotypem připomínají metabolický syndrom lidí s viscerální obezitou, dyslipidemií, inzulinorezistencí a hypertenzí. Naopak myši s knock-outovaným genem pro 11-β-HSD1 jsou vitální, zdravé, mají kardioprotektivní lipidický profil se zvýšeným HDL-C, zvýšeným apoproteinem A1, sníženou hladinou triglyceridů, vykazují zvýšenou citlivost na inzulin a zlepšenou glukosovou toleranci.Tyto myšky na dietě s vysokým obsahem tuků odolávají váhovému přírůstku. Jediným nalezeným potencionálním negativem u 11-β-HSD1 -/- myši je hyperplazie nadledvin jako projev zvýšené aktivity osy HHA kompenzující zvýšený periferní obrat kortisolu tak,aby byla zachována jeho normální plazmatická koncentrace. Dalším krokem v poznání byly studie na lidech, které srovnávali štíhlé a obézní lidi, většina těchto studií prokázala zvýšenou expresi a aktivitu 11-β-HSD1 v tukové tkáni (subkutánní i viscerální) obézních jedinců, byly nalezeny těsné pozitivní korelace mezi zvýšením 11-β-HSD1 na straně jedné a BMI, obvodem pasu a objemem viscerální tukové tkáně na straně druhé. Zvýšená produkce kortisolu viscerálním tukem, která může představovat až ¼ cirkulujícího kortisolu, je přiváděna ze splanchnické oblasti portální žílou do jater, kde významně přispívá k hepatální inzulinorezistenci, protože zvyšuje glukoneogenezi a výdej glukosy játry.
Na první pohled překvapivý je zjištění, že aktivita 11-β-HSD1 v játrech obézních je snížená, zřejmě se jedná o kompenzační mechanismus, který má za úkol bránit dalšímu rozvoji obezity, snížit inzulinorezistenci, snížit hepatální výdej glukosy a udržet tak normální glukosovou toleranci. Na druhou stranu u obézních diabetiků 2. typu tento mechanismus selhává.
Exprese 11-β-HSD1 byla prokázána také v Langerhansových ostrůvcích pankreatu. Takto lokálně vznikající kortisol auto či parakrinně blokuje sekreci inzulinu beta-buňkami. Zvýšená exprese a aktivita 11-β-HSD1 v Langerhansových ostrůvcích proto nepochybně hraje významnou úlohu v manifestaci a progresi diabetu.
V tukové tkáni obézních bylakromě zvýšené exprese 11-β-HSD1 zjištěna i zvýšená exprese receptorů pro glukokortikoidy což zvětšuje lipolytický účinek kortisolu na adipocyty zvýšením aktivity hormon-senzitivní lipázy. Dochází ke zvýšenému uvolňování volných mastných kyselin a chronickému vzestupu jejich plazmatické koncentrace s následnými projevy tzv. lipotoxicity v řadě orgánů:
- Játra. Zvýšený přísun MK do buněk zvyšuje glukoneogenezi a syntézu triglyceridů. Zvýšená glukoneogeneze a výdej glukosy z jater zvyšuje glykemii a zhoršuje glukosovou toleranci. Triglyceridy vytvořené hepatocyty se mohou v játrech hromadit pod obrazem jaterní steatózy s rizikem přechodu do steatofibrózy až cirhózy.
- Kosterní svaly. Zvýšená nabídka mastných kyselin snižuje vychytávání glukosy svaly a tím zhoršuje celkový stav inzulinorezistence.
- Cévy. Hromadění lipidů v jejich stěně je kritickým bodem patogeneze aterosklerotického procesu vedoucího k následným kardiovaskulárním komplikacím typu ICHS a CMP.
- Myokard. Akumulace triglyceridů v kardiomyocytech snižuje čerpací schopnost srdce, což může mít v kombinaci s postižením koronárních tepen fatální následky.
- Pankreas. Hromadící se tuk přímo poškozuje β-buňky Langerhansových ostrůvků, jednak funkčně snížením jejich citlivosti a tím snížení sekrece inzulinu, jednak organicky zvýšením jejich apoptózy.
- Ledviny jsou lipotoxicitou poškozeny jednak z důvodu aterosklerózy svých cév, jednak poškozením exkreční funkce tubulů, což nepochybně souvisí s častým výskytem arteriální hypertenze u obézních.
Inhibice 11-β-HSD
Po přečtení výše uvedeného není s podivem, že blokáda účinku 11-β-HSD se stala potencionální terapeutickým cílem v léčbě obezity, cukrovky 2. typu a metabolického syndromu. To vedlo zejména v poslední dekádě k nebývalé snaze farmaceutických firem o nalezení takového léku. Jaké jsou výsledky? Nalezené látky si rozdělíme dle mechanismu a rozsahu jejich účinku:
- Neselektivní inhibitory 11-β-HSD blokují oba typy enzymu, jenže žádoucí je pouze blokáda 11-β-HSD1, protože blokáda účinku 11-β-HSD2 vede k nežádoucím účinkům v podobě volumové hypertenze a hypokalemie. Mezi tyto látky patří již dlouho známá kyselina glycyrrhetinová, která je obsažena v lékořici. Jiným neselektivním inhibitorem je karbenoxolon, který je v inhibici méně potentní než kys.glycyrrhetinová, což znamená i menší ovlivnění tlaku a hladiny draslíku, ovšem současně bohužel zdá se blokuje převážně aktivitu 11-β-HSD1 v játrech a méně v tukové tkáni, což má u obézních za následek, že sice dojde ke zlepšení plazmatického lipidového profilu případně jaterní steatózy, zlepší se inzulinová senzitivita snížením jaterní glukoneogeneze a výdeje glukosy játry, ale nedochází ke snížení příjmu stravy a snížení hmotnosti.
- Selektivní inhibitory 11-β-HSD mají mnohonásobně vyšší afinitu k 11-β-HSD1 než 11-β-HSD2 a proto postrádají výše uvedené nežádoucí účinky na krevní tlak a kalemii. Biochemicky jde o pestrou skupinu látek odvozených od sulfonamidu, thiazolonu, carboxamidu či triazolu. V současné době jsou tyto látky v 1. a 2. fázi klinických studií . Výsledky jsou nadějné, je popisováno snížení inzulinorezistence, snížení glykemie nalačno, snížení hladiny glykovaného hemoglobinu, zlepšení lipidového profilu a u některých i snížení tělesné hmotnosti.Jaké můžeme očekávat a jaké skutečně jsou nežádoucí účinky této skupiny inhibitorů? Ze studií na 11-β-HSD1 -/- myší víme, že dochází ke kompenzatorní aktivaci osy HHA, zvýšení sekrece ACTH, hyperplazii kůry nadledvin a zvýšení sekrece kortisolu, které zabrání poklesu plazmatické hladiny kortisolu při jeho snížené periferní „recyklaci“. Studie na lidech ukázali, že k této aktivaci dojde, ale hladiny ACTH se zvedají cca jen dvojnásobek a i po tomto zvýšení většinou zůstávají v mezích normy, plazmatické koncentrace kortizolu jsou fyziologické a zachovávají si cirkadiální rytmus, po vysazení léku dochází k normalizaci ACTH. Z toho tedy můžeme zatím usuzovat, že aktivace osy HHA při použití selektivních blokátorů 11-β-HSD1 je mírná, kompenzace není přemrštěná (nedochází k projevům hyperkorticismu) a je plně reverzibilní. Zvýšená hladina ACTH ovšem nestimuluje jen sekreci kortisolu, ale také nadledvinových androgenů, což by mohlo vést u léčených žen k projevům hirsutismu a poruch menstruačního cyklu. Zvýšená hladina ACTH v menší míře než v případě kortisolu a nadledvinových androgenů zvyšuje i tvorbu aldosteronu, což by mohlo vést k projevům primárního hyperaldosteronismu jako je hypertenze a hypokalemie. I tyto možnosti již byly studovány. Výsledky neukázaly zvýšení ani hladiny aldosteronu a ani změny elektrolytů, které by odpovídaly zvýšené mineralokortikoidní aktivitě. V protikladu k tomu, ale byly pozorovány zvýšené koncentrace DHEA a androstendionu u obou pohlaví a vyšší hladiny testosteronu u žen (ovšem bez signifikantního zvýšení indexu volných androgenůpo zohlednění koncentrace SHBG). Klinicky se tyto mírně zvýšené hladiny neprojevily, ale studie byly příliš krátké na dostatečné posouzení, i když většina žen pravděpodobně nebude mít vzhledem k nevelké elevaci androgenů, u menší citlivější skupiny nelze problémy vyloučit.
- Kompetitory 11-β-HSD jsou látky strukturálně podobné glukokortikoidům se kterými soutěží o 11-beta-HSD a tím oslabují její účinnost.Patří sem například 7-hydroxy-DHEA či 7-oxo-DHEA, který je endogenním steroidem, ale na rozdíl od DHEA nemá androgenní účinnost, zato u něho byly popsány účinky imunomodulační a termogenní.
Kortison reduktázová deficience (CRD)
Jde o dědičné onemocnění způsobené mutací genu pro 11-β-HSD1 nebo mutací genu pro H6PD . Funkčním následkem těchto mutací je snížená aktivita 11-β HSD, nedostatečná přeměna kortisonu na kortisol a tím i zvýšený obrat kortisolu v periferních tkáních. Zvýšená spotřeba kortisolu vede mechanismem negativní zpětné vazby k aktivaci hormonální osy hypotalamus-hypofýza-kůra nadledvin, hormonálním důsledkem je zvýšená sekrece ACTH z adenohypofýzy, což postačuje k udržení normální plazmatické koncentrace kortisolu, ale dochází ke zvýšení hladin nadledvinových androgenů (DHEA, androstendion). Vzniklá hyperandrogenemie má u postižených žen za následek rozvoj hirsutismu s poruchami menstruačního cyklu ve smyslu oligmenorhey až amenorey spojené s anovulací a poruchou fertility. Příznaky se objevují v období dospívání a rané dospělosti. U postižených mužů jsou symptomy méně nápadné, obvykle pod obrazem pubarchae praecox, tedy jako předčasný vývoj pubického a/nebo axilárního ochlupení u hochů před 9. rokem života. Biochemickým markerem CRD je snížený poměr metabolitů kortisolu vůči metabolitům kortisonu v moči. Bez ohledu na pohlaví postižení nemají projevy hypokorticismu, zvýšený obrat kortisolu je vyvážen jeho zvýšenou sekrecí, nehrozí jim proto rozvoj adrenokortikální krize, který známe např. od pacientů s těžkými formami kongenitální adrenální hyperplazie. Terapeuticky lze využít syntetický kortikoid dexamethason, který útlumem sekrece ACTH sníží zvýšenou hladinu nadledvinových androgenů a zlepší tak potíže pacientů.
Pseudohyperaldosteronismus z nedostatečné aktivity 11-β-HSD2
Pseudohyperaldosteronismus je skupina stavů, které klinicky a laboratorně připomínají primární hyperaldosteronismus (Connův syndrom, adenom kůry nadledvin s nadprodukcí aldosteronu), ale při tom mají normální či snížené hladiny aldosteronu. Klinickým příznakem je nález volumové hypertenze, laboratorně nacházíme hypokalemii, metabolickou alkalózu, nízkou hladinu aldosteronu i reninu, vzácně hypernatremii. Hypokalemie může vést k projevům svalové slabosti až rhabdomyolýzy a také ohrožuje pacienta srdeční arytmií.
Mezi pseudohyperaldosteronismy patří:
(1) vzácnější forma kongenitální adrenální hyperplazie (CAH) s mutací genu pro 11-β-hydroxylázu, výsledkem je nedostatečná syntéza kortisolu, která zvyšuje sekreci ACTH a to vede k hromadění steroidů před enzymatickým blokem, což kromě nadledvinových androgenů zahrnuje u tohoto bloku i 11-deoxykortikosteron, mineralokortikoidně působící prekurzor aldosteronu, výsledkem je virilizace genitálu u děvčátek, předčasná puberta u chlapců a u obou pohlaví hypertenze s hypokalemií.
(2) Liddleův syndrom, což je autozomálně dominantně dědičná mutace epiteliálního sodíkového kanálu (ENaC) hlavních buněk distálních tubulů a sběracích kanálcích, jde o mutaci aktivační, takže dochází k nekontrolovaně vysoké resorpci sodíku bez vlivu aldosteronu a k rozvoji hypertenze.
(3) Snížená aktivita 11-β- HSD2, která má za následek, že i běžné nezvýšené hladiny kortisolu mají mineralokortikoidní efekt, selhává ochrana mineralokortikoidních receptorů před glukokortikoidy. (a) Příčina snížené aktivity 11-β HSD2 může být vrozená jako v případě syndromu zdánlivého nadbytku mineralokortikoidů (apparent mineralocorticoid excess = AME), což je velmi vzácná autozomálně recesivně dědičná mutace genu pro 11-β-HSD2. (b) Aktivita 11-β-HSD2 může být snížena i z příčin získaných, příkladem je intoxikace lékořicí, jejíž extrakty se používali zejména dříve pro své expektorační, antiflogistické a gastroprotektivní účinky. Lékořice obsahuje kyselinu glycyrrhiziovou, která neselektivně blokuje 11b-hydroxysteroidní dehydrogenázu.
(4) V širším slova smyslu můžeme mezi pseudohyperaldosteronismy zařadit i Cushingův syndrom (zvýšená endogenní produkce kortisolu nebo zvýšený přísun exogenních kortikoidů). Zde se jedná o relativní insuficienci 11-β-HSD2, kdy kapacita tohoto enzymu nezvládá konvertovat a deaktivovat příval kortikoidů v ledvinách při jejich vysokých plazmatických koncentrací a tyto pak zahlcují mineralokortikoidní receptory.
Ať už je příčina pseudohyperaldosteronismu jakákoliv výsledkem je volumová hypertenze, nikoliv edémy. Proč? Určitá retence sodíku a s ní spojená volumoexpanze nastane, ale to má za následek nárůst krevního tlaku, což vede mechanismem tlakové diurézy ke zvýšení výdeje sodíku resp. k obnově rovnováhy mezi příjmem a výdejem Na+. Hypertenze zde představuje daň, kterou zaplatí organismus za obnovu bilance sodíku. Vysoký krevní tlak je ledvinami použit jako „nástroj“ k vyloučení soli, jako „oběť“ nutná k vyvážení zvýšené retence sodíku, která nastala následkem insuficience 11-β-HSD2 či jiné příčiny pseudohyperladosteronismus. Arteriální hypertenze je z pohledu přežití organismu podstatně menší problém než nerovnováha v příjmu soli a vody, která by během několika hodin až dnů vedla k otoků a volumovému přetížení srdce s jeho následným selháním.
Použitá literatura
- BIČÍKOVÁ M., STÁRKA L. Inhibice 11-β-hydroxysteroiddehydrogenázy typu 1 jako možná cesta léčby diabetu, obezity a metabolického syndromu. DMEV, 2011, 14(2): 61-66
- BOSCARO M, GIACCHETTI G, RONCONI V. Visceral adipose tissue: emerging role of gluco- and mineralocorticoid hormones in the settings of cardiometabolic alterations. Ann.N.Y.Acad.Sci.,2012,1264: 87-102
- Di DALMAZI G., PAGOTTO U., PASQUALI R. VICENNATI V. Glucocorticoids and type 2 diabetes: from fysiology to patology. J. of Nutrition and Metabolism, 2012, 2012:525093
- DRAPER N., STEWART PM. 11-β-hydroxysteroid dehydrogenase and the pre-receptor regulativ of corticosteroid hormone action. J. of Endocrinology, 2005, 186(2): 251-271
- HOLLIS G., HUBER R. 11-β-hydroxysteroid dehydrogenace type 1 inhibition in type 2 diabetes mellitus. Diabetes, Obesity and metabolism, 2010,13(1): 1-6
- JOHARAPURKAR A, DHANESHA N, SHAH G et al. 11-β-hydroxysteroid dehydrogenace type 1: potential therapeutic target for metabolit syndrome. Pharmacological reports, 2012, 64(5): 1055-1066
- LAWSON AJ, WALKER EA, LAVERY GC et al. Cortisone-reductase deficiency associated with heterozygot mutations in 11-β-hydroxysteroid dehydrogenace type 1. PNAS, 2011, 108(10): 4111-4116
- PAULSEN SK, PEDERSEN SB, FISKER S, RICHELSEN B. 11-β-HSD type 1 expression in human adipose tissue: Impact of gender, obesity and fat localization.Obesity(Silver Spring), 2007, 15(8): 1954-60
- PRASAD SAKAMURI SS., SUKUPAKA M., PRATHIPATI VK et al. Carbenoxolone treatment ameliorated metabolit syndrome in WNIM/Ob obese rats, but induced severe fat loss and glukose intolerance in lean rats. PLoS One, 2012, 7(12): e50216
- ROUBALOVÁ J. Kortizol a jeho účast na regulaci energetického metabolismu.Bakalářská práce studijního oboru biologie, UK Praha, 2009.
- VALSAMAKIS G, ANWAR A, TOMLINSON W et al. 11-β-hydroxysteroid dehydrogenace type 1 activity in lean and obese males with type 2 diabetes mellitus. J. of clinical endokrinology and metabolism, 2004, 89(9):4577-4761
- WAKE DJ., WALKER BR. 11-β-hydroxysteroid dehydrogenace type 1 inhibition in obesity and the metabolic syndrome. Molecular and Cellular Endocrinology, 2004, 215(1-2): 45-54
 metabolické poruchy")

