

Charakteristika
Hemofilie znamená krvácivost, a označuje tedy zvýšenou tendenci ke krvácení. Může vzniknout akutně , ale dobře známé jsou chronické formy. Mezi nimi jsou zastoupeny vrozené i získané krvácivé stavy.
Vůbec nejčastější vrozenou krvácivou nemocí je von Willebrandova choroba. Klasické případy hemofilie jsou spojeny také s abnormalitami faktorů přiřazovaných vnitřní cestě aktivace krevního srážení.
Vrozené krvácivé poruchy provázející deficit faktorů vnitřní cesty aktivace hemokoagulační kaskády
Porucha při deficitu faktoru XII
Pacienti s deficitem faktoru XII mají prokazatelnou poruchu aktivace koagulace vnitřní cestou – prodloužený PTT. Netrpí ale ani spontánním krvácením ani zvýšenou krvácivostí při úrazech anebo chirurgických výkonech. Mírnost těchto projevů zpochybňuje klasické postavení faktoru XII jako činitele aktivujícího vnitřní cestu koagulace.
Porucha při deficitu faktoru XI (hemofilie C)
Krvácivost z deficitu faktoru XI dědičného autosomálně recesivně se projevuje snadnějším anebo delším krvácením po traumatech anebo po chirurgických výkonech (např. po extrakci zubu). Krvácení je více vyjádřeno ve tkáních, kde je vysoká aktivita fibrinolytického systému. Klinický průběh je mírnější než u hemofilie A či B.
Rozdíl mezi závažností poruchy při deficitu faktoru XI ve srovnánís deficitem faktoru XII je vysvětlen skutečností, že že faktor XI je aktivován i jinými cestami než jen faktorem XII. Jedním z významných způsobů aktivace faktoru XI například je pozitivní zpětnovazební smyčka aktivovaným trombinem.
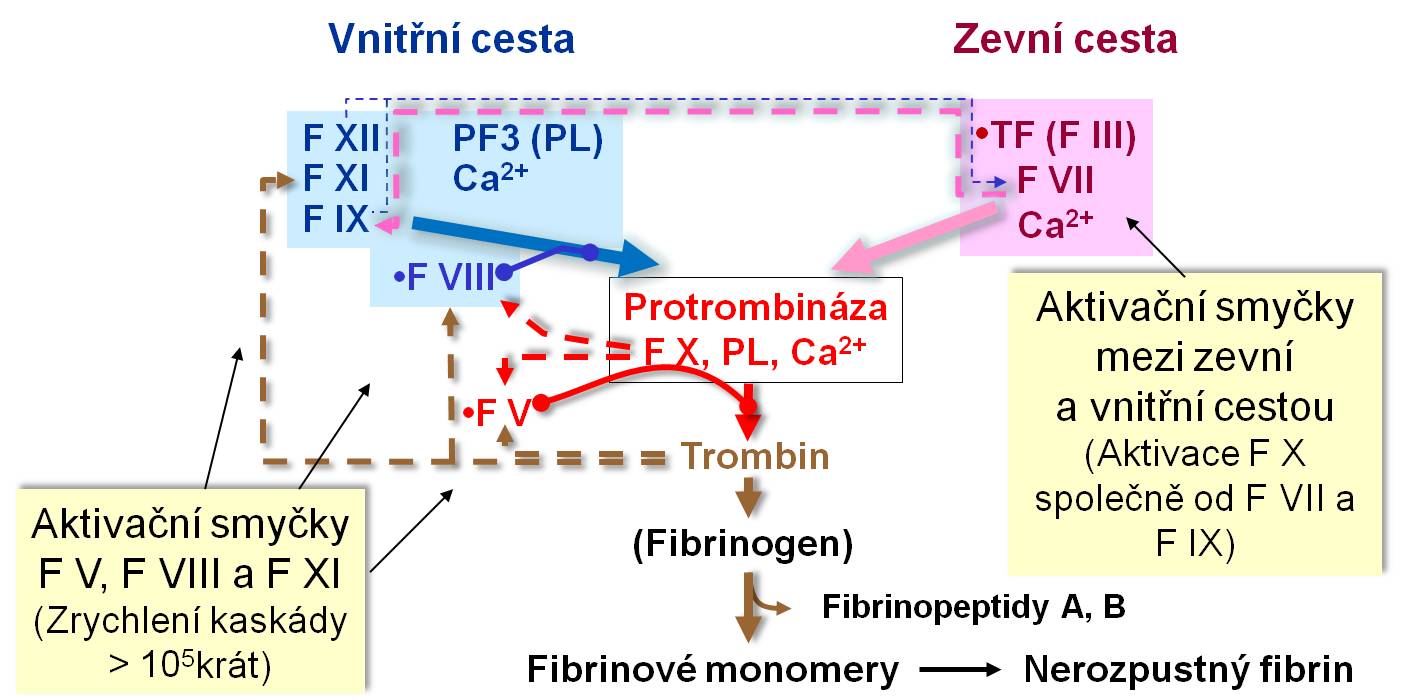
Obr. 1. Aktivační zpětnovazební smyčky v koagulační kaskádě. Schéma je možno zhlédnout i v animované podobě – za prvé, aktivační smyčky mezi zevní a vnitřní cestou, a za druhé, aktivační smyčky FV, FVIII a FXI spolu s funkcí FXIII a účastí kalikrein-kininového systému.
Porucha při deficitu faktoru VIII a IX (hemofilie A a B)
Při deficitu kteréhokoliv z obou zbývajících faktorů přiřazovaných vnitřní cestě, tzn. faktoru IX anebo faktoru VIII, vznikají těžké formy hemofilie charakterizované spontánním krvácením do kloubů, svalů a měkkých tkání. Taková krvácení mohou znamenat ohrožení života. Onemocnění vzniklé deficitem faktoru VIII se označuje jako hemofilie A, deficitem faktoru IX pak hemofilie B. Obě tyto formy se dědí gonosomálně recesivně.
Dramatický rozdíl příznaků a závažnosti krvácení při hemofilii A a B ve srovnání s daleko mírnějšími příhodami anebo poruchami provázejícími deficit faktorů XII a XI, na nichž je aktivace faktorů IX a VIII závislá, je na první pohled překvapující. Je však vysvětlen tím, že faktor IX je významně aktivován aktivním komplexem TF-F VII a spolu s ním se pak účastní aktivace faktoru X (viz obrázek). Navíc je zpětnovazební smyčkou trombinem aktivován kofaktor F IX faktor VIII (viz obrázek). Při deficitu faktoru IX (hemofilie B) se pozitivní amplifikační signál z F VII nerealizuje, takže aktivace faktoru X je výrazně oslabena. Totéž nastává při deficitu faktoru VIII (hemofilie A), na němž je účinek faktoru IX kriticky závislý.
Von Willebrandova choroba
Von Willebrandova choroba je vůbec nejčastějším vrozeným krvácivým stavem. Projevuje se sklonem ke snadnému krvácení. Krvácivé projevy jsou nejvíce zřetelné ve tkáních protkaných drobnými cévami (podkoží, nosní sliznice, sliznice GIT, větší menstruační krvácení atd.). Kromě vrozených forem však existují i získané formy von Willebrandovy choroby.
Von Willebrandův faktor je kofaktor s širokými vazebnými schopnostmi funkcí bez enzymové aktivity. Funguje organizován do multimerů. Účastní se 1. i 2. fáze hemostázy.
- V první fázi hemostázy jednak ulpívá na obnažených vláknech kolagenu, jednak váže řadu proteinů (receptorů) přítomných na povrchu trombocytů, takže slouží jako organizační centrum pro adhezi a agregaci destiček.
- Druhé fáze hemostázy se zejména účastní jako ochranný protein faktoru FVIII. Neaktivní FVIII v plazmě je v komplexu s von Willebrandovým faktorem chráněn před degradací. Význam FVIII v koagulační kaskádě byl připomenut výše.
Defekty anebo deficit von Willebrandova proto vedou k poruše adheze a agregace trombocytů a ke snížení aktivity FVIII. Funkční defekty jsou závislé na povaze a poloze mutací von Willebrandova faktoru. To vysvětluje, proč lze von Willebrandovu chorobu dále klasifikovat do subtypů.
Získané formy von Willebrandovy choroby souvisejí s poruchami jeho inaktivace. Za normálních podmínek jsou multimery von Willebrandova faktoru v plazmě inaktivovány specifickými proteázami (např. metaloproteázou ADAMTS13, a disintegrin-like and metalloprotease thrombospondin type 1 motif no. 13). V některých případech, např. zvýšeným smykovým třením v tenkých cévách anebo při stenóze aortální chlopně, může dojít k destabilizaci multimerů von Willebrandova faktoru a k obnažení kritických míst v jeho struktuře. Zvýšené štěpení von Willebrandova faktoru proteázami následně vede ke krvácivým projevům získané formy onemocnění.
Naproti tomu nedostatečná funkce specifických proteáz, které inaktivují von Willebrandův faktor, způsobí zvýšenou agregaci destiček a tvorbu fibrinu v malých cévách provázené mikroangiopatickou hemolytickou anémií. Ucpání cév mikrotromby může vést k nekróze kapilár s následným rozvojem purpury. Tyto poruchy se vyskytují:
- Při hemolyticko-uremickém syndromu, kdy v popředí orgánového postižení jsou ledviny;
- Při trombotické trombocytopenické purpuře, kdy v popředí orgánového postižení je mozek.
Osoby s krevní skupinou O mají nižší hladiny von Willebrandova faktoru v plazmě než osoby se skupinami A, B a AB. Vyšší aktivita von Willebrandova faktoru souvisí s vyšším rizikem trombóz a ischemických příhod (nejvyšší u osob se skupinou AB).
Zásady terapie akutních krvácivých stavů
Při krvácivých stavech pacienta je indikováno podání krevních derivátů anebo koncentrátů prokoagulačních faktorů.
Zpracoval: Jaroslav Veselý, Ústav patologické fyziologie LF UP
