

Úvod
Diabetes mellitus neoznačuje jedinou nemoc. Je to název pro velkou skupinu onemocnění. Všechna tato onemocnění společně charakterizuje hyperglykémie, k níž dochází následkem poruch sekrece inzulínu, působení inzulínu anebo následkem kombinace obou těchto příčin.
Rozhodovací mezí pro diagnózu diabetu je hodnota plazmatické glukózy nalačno rovna nebo větší než 7,0 mmol/l (FPG, fasting plasma glucose, ≥ 7 mmol/l). Při této hranici začíná strmý růst výskytu závažmých pozdních kompikací diabetu (obr.1).

Obr. 1. Prevalence výskytu mikroangiopatií v závislosti na koncentraci plazmatické glukózy měřené nalačno. Je patrný prudký nárůst chronických komplikací způsobených hyperglykémií nad 6,9 mmol/l. (Podle: Diab Care 2002 25:s5-s20.)
Diabetes je nebezpečné onemocnění, a to jak pro své závažné chronické následky, tak pro své těžké akutní komplikace. Chronická hyperglykémie způsobuje poškození všech orgánů a tkání, zejména:
- Cév (makro- a mikroangiopatie provázené poruchou drenážních a nutričních funkcí mikrocirkulace);
- Ledvin (glomeruloskleróza);
- Nervového systému (neuropatie somatického i autonomního nervstva);
- Očí (retinopatie, katarakta).
Diabetes je pátou nejčastější příčinou úmrtí se strmě narůstající epidemickou incidencí. Většinu osob s diabetem tvoří obézní.
Pacienti trpící diabetem mají 2 – 4 x vyšší riziko kardiovaskulárního onemocnění než osoby bez diabetu.
Diabetes je dopovědný za asi 40 % všech nových případů terminálního postižení a selhání ledvin.
Diabetes je odpovědný za největší část případů slepoty, k níž odchází mezi 20 – 75 roky života.
Dlouhodobými následky diabetu také jsou:
- Dyslipidémie (diabetes je významným rizikovým faktorem aterosklerózy);
- Hypertenze;
- Nemoci periodontu;
- Psychologické problémy;
- Sociální problémy.
Diabetes ovšem také provázejí těžké až smrtící akutní kompikace:
- Hyperglykemický hyperosmolární syndrom, hyperglykemické hyperosmolární kóma;
- Hyperglykemické hyperosmolární ketoacidotické kóma;
- Laktacidóza, laktacidotické kóma;
- Hypoglykémie, hypoglykemické kóma.
Etiopatogenetické základy klasifikace diabetu
Diabetes vuniká následkem následkem poruch sekrece inzulínu, působení inzulínu anebo následkem kombinace obou těchto příčin. Inzulín hlavně působí na tři cílové tkáně: jaterní, svalovou a tukovou. Pro vznik hyperglykémie je rozhodující ztráta účinků inzulínu na játra. Játra, postrádající při diabetu inhibiční vliv inzulínu na glukoneogenezu, neřízeně produkují velká množství glukózy do krevního oběhu. Příspěvek snížené spotřeby glukózy v ostatních periferních tkáních k hyperglykémii při diabetu je ve srovnání se zvýšenou sekrecí glukózy z jater druhořadý.
Podle etiopatogeneze lze většinu diabetických onemocnění zařadit do dvou širokých skupin:
Skupinu diabetu 1. typu, kdy příčinou onemocnění je snížení až vymizení sekrece inzulínu z β-buněk pankreatu;
Skupinu diabetu 2. typu, kdy produkce inzulínu je dlouhou dobu zachována, ale za onemocnění je odpovědná porucha citlivosti tkání k inzulínu (rezistence k účinkům inzulínu), která nemůže být kompenzována patřičným nárůstem sekrece inzulínu z β-buněk pankreatu.
Zvláštní kategorií je diabetes mellitus těhotných (gestační diabetes mellitus). Je vhodné mít na paměti, že k částečnému zhoršení tolerance glukózy v těhotenství dochází i fyziologicky, ale obvykle až ve třetím trimestru.
Podrobná klasifikace diabetu je k otevření zde.
Diabetes 1. typu
Při diabetu 1. typu primárně dochází k poškození a zániku β-buněk pankreatu. Osob postižených diabetem 1. typu je mnohem méně než těch, které trpí diabetem 2. typu. Za destrukci β-buněk je zpravidla odpovědný agresivní autoimunitní proces. Identifikuje se na základě zjištění přítomnosti autoprotilátek. Někdy se imunitní povahu procesu nepodaří prokázat, a potom se hovoří o idiopatickém diabetu 1. typu.
Protilátky zjišťované v plazmě nemocných jsou zaměřeny proti inzulínu (IAA, insulin autoantibodies), proti cytoplazmě ß-buněk pankreatu (ICA, islet cell autoantibodies), proti dekarboxyláze kyseliny glutamové (anti-GAD65, anti-glutamic acid decarboxylase) anebo proti izoformám tyrosinfosfatázy (ostrůvkový antigen, islet antigen IA-2A, IA-2ßA).
Autoimunitní diabetes 1. typu obvykle začíná v dětství anebo v pubertě. Může se však vyskytnout v kterémkoliv, a to i pokročilém věku (v 8. anebo 9. dekádě života). Pacienti obvykle nejsou obézní, ale obezita je s touto diagnózou slučitelná. Destrukce buněk může být velmi rychlá (obvykle v raném věku), nebo postupovat pomalu (obvykle u dospělých). Genetická složka není silná, i když statisticky existují vazby (protektivní nebo škodlivé) v systému HLA-DR/DQA,DQB. Imunitní diabetes 1. typu se nezřídka sdružuje s jinými autoimunitními onemocněními (sdružené autoimunity).
Latentní autoimunitní diabetes dospělých (LADA) často zpočátku bývá klasifikován jako diabetes 2. typu. Až u 80 % trpících diabetem LADA však jsou přítomny přítomny autoprotilátky, zejména proti GAD65. Proto se i tato forma diabetu dospělých klasifikuje jako diabetes 1. typu.
Ve vývoji diabetu 1. typu existuje zpravidla krátké skryté (latentní) stádium (prediabetes) charakterizované ještě normální (do 5,6 mmol/l) anebo mírně zvýšenou (5,6 – 6,9 mmol/l) glykémií nalačno při současně už alterované sekreční funkci β-buněk. Toto stádium se klinicky podaří zachytit jen zcela výjimečně. Po kritickém snížení sekreční kapacity β-buněk pro inzulín se pak diabetes manifestuje naplno (FPG ≥ 7,0 mmol/l) (obr. 1). Jak už bylo uvedeno, příčinou hyperglykémie u diabetu je odblokování glukoneogeneze a vysoká produkce glukózy z jater při absenci vlivu inzulínu. Po vymizení inzulínu se v plazmě nedaří zjistit ani inzulín, ani C-peptid.
V nepřítomnosti inzulínu je desinhibována lipolýza v tukové tkáni. Pacienti s diabetem 1. typu proto jsou ve vysoké míře predisponováni ke vzniku ketoacidózy z mastných kyselin; riziko ketoacidózy je mnohem vyšší, než tomu je u pacientů s diabetem 2. typu.

Obr. 2. Změny glykémie a inzulinémie při vzniku diabetu 1. typu. Prohlubující se hypoinzulinémii (absolutní hypoinzulinémii) následuje vzestup plazmatické glukózy. Zprvu se vyvíjí stádium prediabetu (IFG), klinicky zachycené jen výjimečně, pak, po kritickém poklesu produkce inzulínu, nastává manifestní diabetes (FPG ≥ 7 mmol/l).
Diabetes 2. typu
Diabetes 2. typu je mnohem větší skupino a zahrnuje daleko větší počet případů než diabetes 1. typu. Postihuje převážně dospělé. Rozvíjí se plíživě a diagnóza je často pozdní, když už zvýšené hladiny hyperglykémie mohly navodit částečné sekundární změny (angiopatie, neuropatie). Hlavními rizikovými faktory jsou:
- Obezita;
- Nízká fyzická aktivita;
- Věk.
Genetická složka je vyjádřena silněji než v případě diabetu 1. typu, vlohy jsou však komplexní. Je téměř jisté, že existuje větší počet etiologicky odlišných forem tohoto onemocnění.
Primárně jde o vznik inzulínové rezistence v periferních tkáních, kterou zároveň provází defekt sekrece inzulínu z β-buněk. Pro vznik hyperglykémie je rozhodující ztráta vlivu inzulínu na glukoneogenezi a na produkci glukózy z jater.
Podobně jako ve vývoji diabetu 1. typu existuje i ve vývoji diabetu 2. typu počáteční klinicky bezpříznakové stádium, které je stavem mezi normální glukózovou homeostázou a manifestovaným diabetem – prediabetes. Prediabetes se i zde ohlašuje zvýšenými koncentracemi glukózy nalačno mezi 5,6 – 6,9 mmol/l (IFG, impaired fasting glucose; anebo – zejména po zátěži glukózou – IGT, impaired glucose tolerance). V protikladu k diabetu 1. typu však v počátečních stádiích vzniku diabetu 2. typu není hypoinzulinémie, nýbrž koncentrace inzulínu v plazmě jsou zvýšeny (hyperinzulinémie, nikoliv hypoinzulinémie, tedy přinejmenším nikoliv absolutní hypoinzulinémie) (obr. 2). β-Buňky touto zvýšenou produkcí inzulínu překonávají rezistenci jater a ostatních periferních tkání, takže se daří udržovat krevní glukózu pod kritickou hranicí 7 mmol/l.
Po dosažení limitu a využití veškeré rezervní kapacity β-buněk koncentrace inzulínu v plazmě kulminují, a nemohou se už více zvýšit (obr. 2). Inzulín už dále nemůže neutralizovat progredující rezistenci tkání. Dochází k dalšímu vzestupu produkce glukózy játry a FGP nad 7,0 mmol/l, a tedy ke klinické manifestaci diabetu.

Obr. 3. Změny glykémie a inzulinémie při vzniku diabetu 2. typu. Narůstající inzulínovou rezistenci tkání, zejména jater, β-buňky překonávají zvýšenou produkcí inzulínu (hyperinzulinémie), která v počáteční, latentní fázi choroby udržuje FGP v mezích prediabetu (poruchy glukózové tolerance). Po dosažení limitu sekrečních rezerv β-buněk, jejichž funkce rovněž může být porušena, ovšem nastává relativní hypoinzulinémie a nastupuje klinický diabetes (FGP ≥ 7 mmol/l). Všimněte si, že tento okamžik – na rozdíl od diabetu 1. typu – nastává při ještě zvýšených absolutních koncentracích inzulínu v plazmě (hyperinzulinémii).
Z etiopatogenetického hlediska je za velkou část onemocnění diabetes mellitus 2. typu s velkou pravděpodobností primárně odpovědné poškození tkání, a zejména jater, lipidy (lipotoxicita). Tento předpoklad je v souladu s údaji o tom, že hlavním rizikovým faktorem diabetu 2. typu je obezita, a to zejména obezita abdominálního typu. Abdominální tuk je mimořádně citlivý na lipolytické stimuly. Nálož uvolněných lipidů míří portálním oběhem právě do jater (obr. 3 a 4).
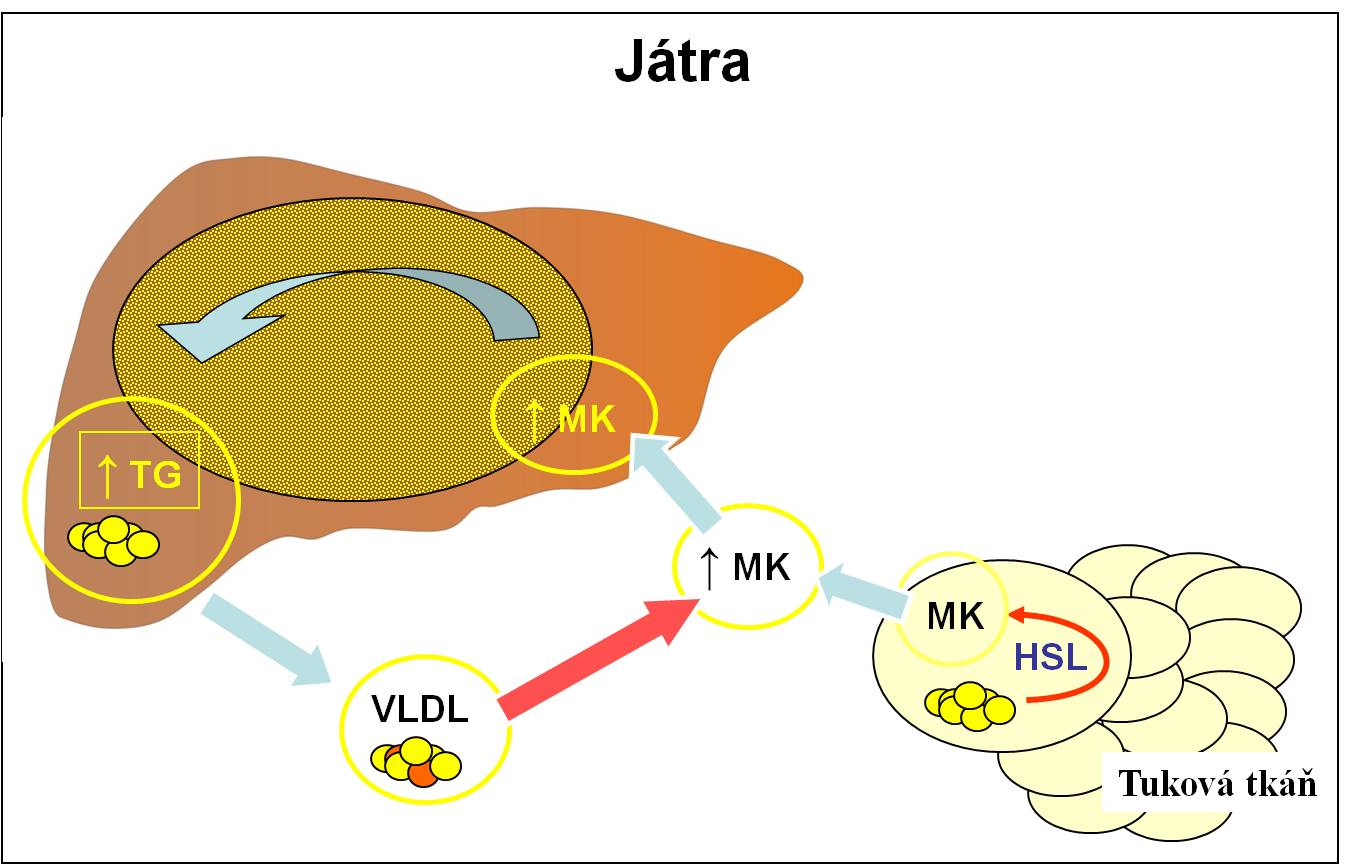
Obr. 3. Schéma lipotoxického působení tuku v játrech. (Podle Browning JD, Horton JD: J Clin Invest 114, 147-152, 2003.)
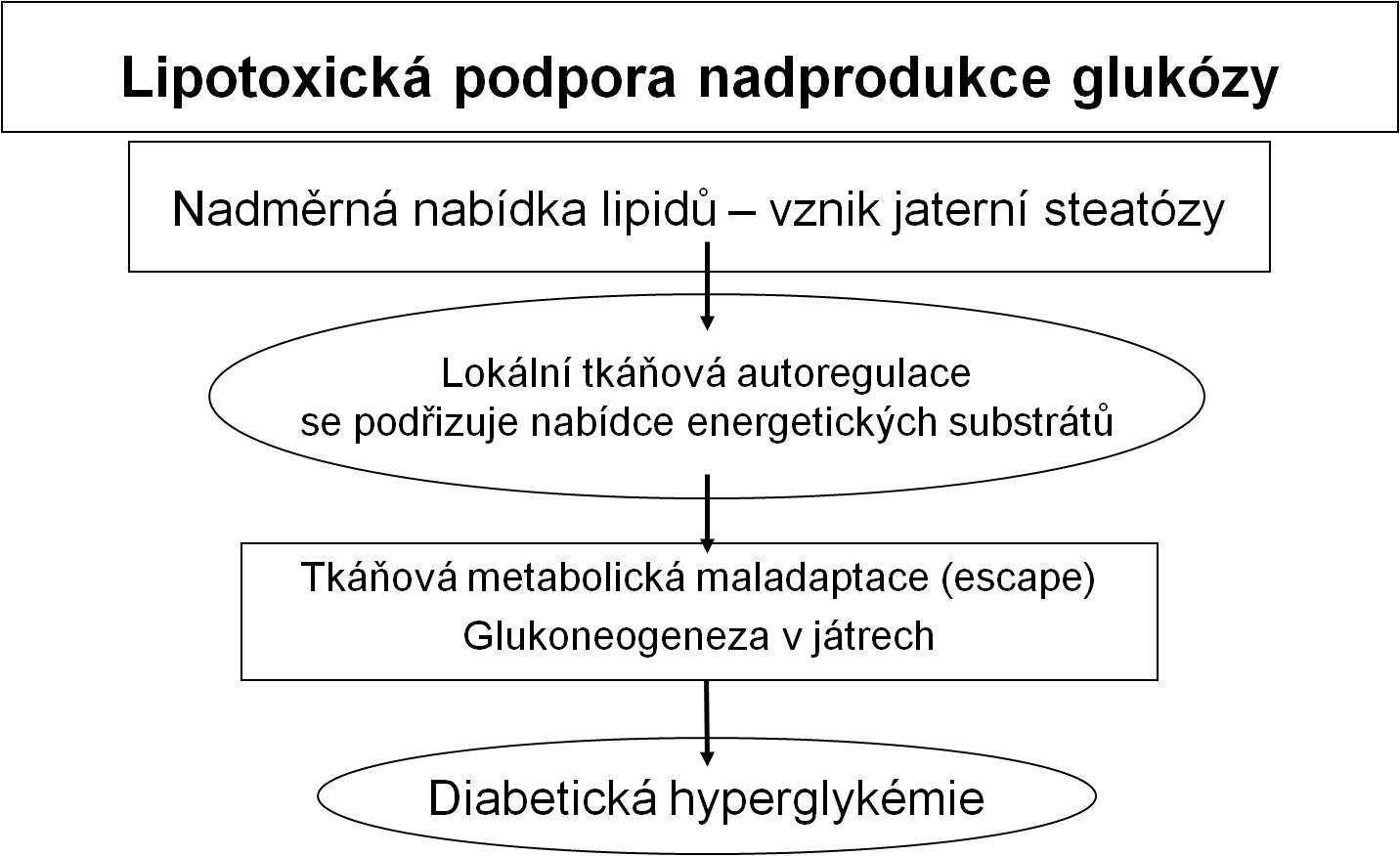
Obr. 4. Schéma uplatnění lipotoxicity při vzniku nadprodukce glukózy v játrech. (Podle Browning JD, Horton JD: J Clin Invest 114, 147-152, 2003.)
Lipotoxicita se kromě toho přímo podílí i na alteraci β-buněk (obr. 5).
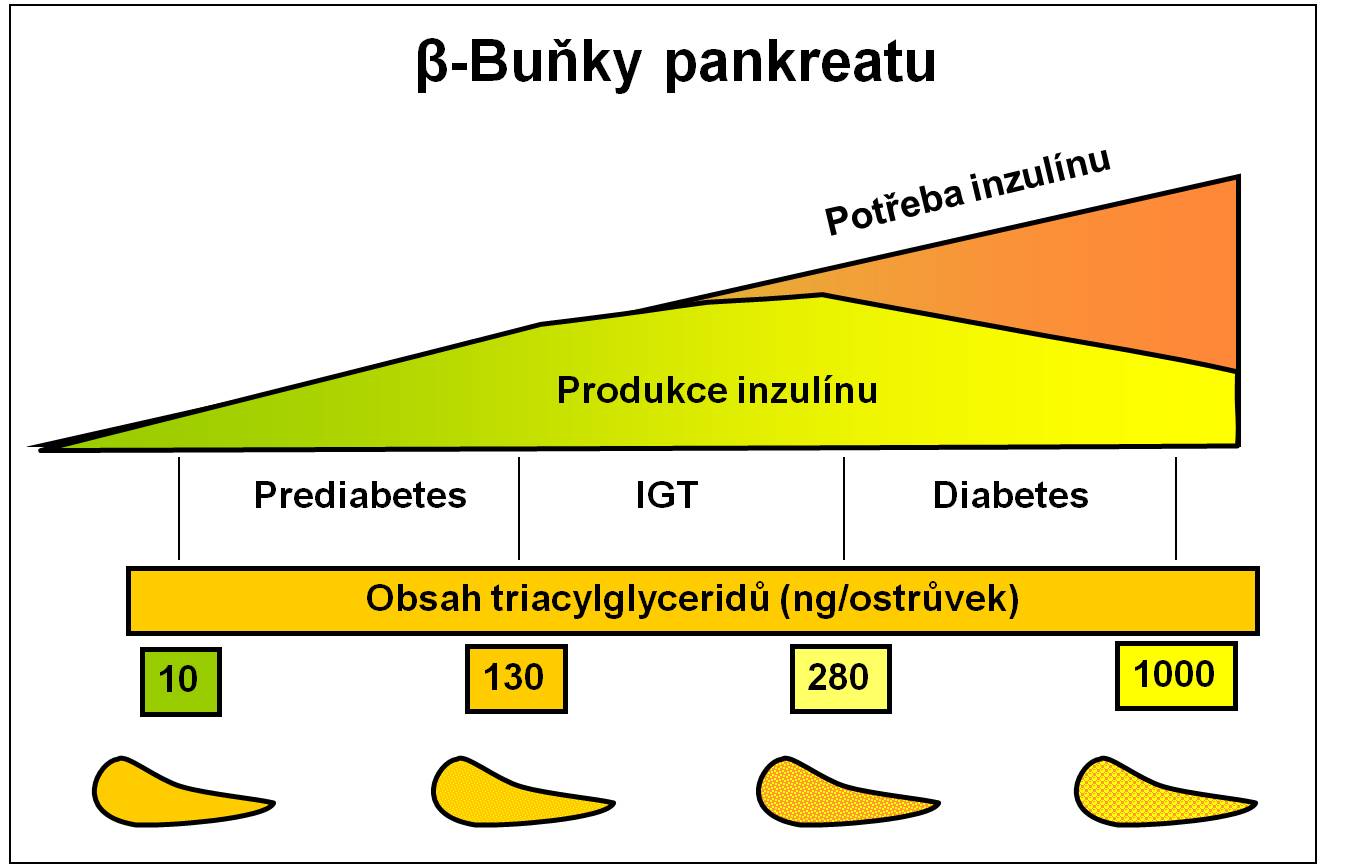
Obr. 5. Ilustrace lipotoxického působení tuku v β-buňkách pankreatu. (Podle Unger RH: Biochimie 87, 57-64, 2005.)
Pacienti s diabetem 2. typu mají spíše relativní, a ne absolutní hypoinzulinémii. Tuková tkáň, která je na účinky inzulínu mimořádně citlivá, proto neuvolňuje vysoká množství mastných kyselin, takže u pacientů s diabetem 2. typu se obvykle vyvíjí ketoacidóza až při těžším stresu (infekce, úrazy, námaha apod.). Zvládnutí ketoacidózy pak ovšem nutně může vyžadovat inzulín, ačkoliv běžně se vystačí s jinou antidiabetickou terapií.
Diabetes mellitus těhotných
Diabetes mellitus těhotných (gestační diabetes mellitus, GDM) postihuje asi 5 % těhotných žen. Zahrnuje jakoukoliv formu poruchy tolerance glukózy, která se poprvé projevila – nebo byla poprve diagnostikována – v průběhu těhotenství.
U většiny těchto případů se po porodu glukózová tolerance vrátí zpátky do normálních podmínek, a žena bude zdravá. Kontroly se provádějí nejdříve po skončení šestinedělí. U malé části budou potíže přetrvávat a žena bude zařazena do jedné ze tří skupin – pacientka s poruchou glukózy nalačno/poruchou glukózové tolerance, s diabetem 1. typu, nebo s diabetem 2. typu.
Gestační diabetes mellitus je závažnou komplikací těhotenství. Zvyšuje perinatální morbiditu a mortalitu. Může být spojen s hypertenzí. Je rizikovým faktorem preeklampsie a eklampsie. Často si vynutí ukončení těhoteství císařským řezem. Rizika je možno omezit náležitou těhotenskou a prenatální péčí, která v nutných případech zahrnuje i podávání inzulínu.
Patofyziologické a klinické aspekty chronických komplikací diabetu
Etiopatologickým podkladem chronických komplikací diabetu jsou nejméně čtyři skupiny procesů souvisejících s abnormálním metabolismem sacharidů:
- Neenzymové glykace proteinů a dalších makromolekulárních struktur. Glykace mění strukturní, funkční i imunologické vlastnosti povrchů a vyvolávají degenerativní změny buněk a tkání. Změněné povrchy se stávají terčem imunologické odpovědi (tvorba a ukládání imunokomplexů, cytotoxické napadení).
- Nadměrná tvorba polyolů z glukózy. Polyoly mění fyzikální, osmotické a funkční vlastnosti sturktur, v nichž se hromadí.
- Nadměrná tvorba hexosaminů.
- Autooxidace glukózy vedoucí ke vzniku reaktivních kyslíkových částic a reaktivních dikarbonylů (oxidační stres, karbonylový stres).
Tyto procesy jsou podkladem následujících závažných změn.
Cévní systém
- Makroangiopatie. Diabetes mellitus patří k hlavním rozikovým faktorům aterosklerózy. Ateroskleróza spolu s následnou trombózou pak je nejčastější příčinou onemocnění tepen a kardiovaskulárních a cerebrovaskulárních onemocnění (a úmrtí).
- Těžkou pozdní komplikací diabetu je syndrom diabetické nohy. Vzniká při ischemické chorobě dolních končetin. Diabetická noha není přímým důsledkem mikro-, ale makroangiopatie. Trpí při ní především kapilární nutritivní funkce. Charakterizuje ji vznik ulcerací, nehojících se defektů a infekce a velmi často vede k amputaci.
- Mikroangiopatie. Mikroangiopatie se rozvíjí ve všech tkáních a orgánech. Klinicky je přístupná sledování ve dvou lokalizacích:
- Diabetická retinopatie (případně s makulopatií) zjistitelná vyšetřením očního pozadí.
- Glomerulopatie (glomeruloskleróza), na jejíž rozvoj usuzujeme na základě vyšetření proteinurie. Dokud se proteinurie udržuje v rozmezí mikroalbuminurie (30 – 300 mg/den), považuje se za reverzibilní. Pro pacienta je v tomto stádiu kritické urgentní nasazení účinné léčby a razantní snížení glykémie, aby se zabránilo další progresi změn. Manifestní proteinurie (nad 300 mg/den) svědčí pro nevratné a zhoršující se poškození glomerulů.
Nervový systém
- Somatické nervstvo. Rozvíji se periferní neuropatie s poruchou aferentní složky (hypestézie, parestézie, dysestézie) i motorické složky (svalová slabost, diabetická amyotrofie). Spolu s cévní složkou má podíl na postižení trofických funkcí (syndrom modrého palce – kazuistika, bércové vředy, diabetická noha, amputace prstů anebo končetin, diabetická artropatie, Charcotův kloub apod.)
- Autonomní nervstvo. Autonomní neuropatie se spolu s cévním postižením podílí na kardiovaskulárních, gastrointestinálních a genitourinálních symptomech a sexuální dysfunkci. Je nutno pamatovat zejména na dvě klinické komplikace, které vypývají z poruchy autonomní aferentace při diabetu:
- Bezbolestné (klinicky bezpříznakové) ischemické příhody myokardu u nemocných diabetem;
- Opožděnou signalizaci hrozící hypoglykémie, a z toho plynoucí náchylnost ke snadnějšímu rozvoji těžkých hypoglykemických stavů (hypoglykemického komatu).
Oční čočka
- Akumulace osmoticky aktivních polyolů v oční čočce se podílí na vzniku katarakty.
Klinický příklad pozdních diabetických změn je popsán v kazuistice umístěné na této adrese.
Patofyziologické a klinické aspekty akutních komplikací diabetu
Diabetické hyperglykemické hyperosmolární kóma
Normální hodnotu osmolality plazmy lze odhadnout pomocí rovnice
osmolalita (mmol/l) = 1,83 ([Na+] + [K+]) + [G] +[ U] + [Ex],
kde [ ] označují látkové koncentrace iontů, glukózy (G), urey (U) a exogenních látek (Ex, např. při intoxikacích – alkohol, etylenglykol, léčiva atd.). Normální osmolalita plazmy je přibližně 290 ± 10 mmol/l.
Kritickým faktorem diabetického hyperglykemického hyperosmolárního stavu je hyperglykémie. Koncentrace glukózy může dosahovat až 30 – 45 mmol/l.
Glukóza přechází do glomeruláního filtrátu. Při překročení tubulárního reabsorpčního maxima (tubulární práh) glukóza přechází do definitivní moči. Při tom její vysoká koncentrace v tubulech brání zpětnému vstřebávání vody. Tím navozená osmotická diuréza vede k enormním ztrátám tekutin z těla. Rozvíjí se hypovolémie, hypotenze a akutní selhání oběhu. Kůže – na rozdíl od hypoglykemického kómatu – zpravidla bývá suchá (dehydratace). Bez pomoci tento stav zpravidla končí smrtí.
Zásady terapie hyperglykemického hyperosmolárního stavu
Naprosto prioritním léčebným opatřením v tomto stavu je rehydratace pacienta. Zároveň s infúzí se dodává chybějící inzulín. Inzulín se pravidelně podává s glukózou, aby se předešlo případné hypoglykémii. Spolu s deficitem vody je nezbytné hradit i deficity iontů, zejména Na+ a K+.
Vzhledem k tomu, že jde o hyperosmolární stav, je nutno dbát na prevenci vzniku mozkového edému. Hyperosmolalitu plazmy je nutno snižovat pomalu, ne více než o 5 mmol/l/hodinu. V případě potřeby je namístě přidat do infúze volumové expandéry (mannitol, dextrany apod.). Osmolalitu nelze zvyšovat glukózou (!); glukóza se metabolizuje na CO2 a H2O. Z hlediska osmotických rovnováh je podání roztoku glukózy podáním čisté vody!
Diabetické ketoacidotické hyperosmolární kóma
Toto hyperglykemické hyperosmolární kóma má navíc složku ketoacidotickou. Rozvrat vodního, iontového a energetického hospodářtví je hlubší než při hyperglykemickém neacidotickém stavu. I v tomto případě je ovšem nutno pamatovat, že život pacienta je na prvním místě ohrožen dehydratací a hypovolémií z osmotické diurézy. Navíc ovšem i acidóza sama může být smrtící.
Geneze ketoacidózy
Při kritickém nedostatku inzulínu (absolutním nebo relativním) dochází jednak k nadměrné mobilizaci mastných kyselin z tukové tkáně a jednak k útlumu jejich resyntézy a reesterifikace. Mastné kyseliny v játrech ve zvýšené míře podléhají β-oxidaci, která končí tvorbou acetylkoenzymu A. Acetylkoenzym A by za normálních okolností mohl být spotřebován v Krebsově cyklu, ale při nedostatku inzulínu váznou anaplerotické reakce, jimiž se normálně z glukózy doplňuje k tomu nezbytný prekurzor, oxalacetát. Acetylkoenzym A se v takovém případě zvýšeně využívá pro tvorbu ketokyselin.
Ketokyseliny – kyselina 3-ketomáselná (acetoctová) a kyselina 3-hydroxymáselná (β-hydroxymáselná) se hromadí v plazmě a tělních tekutinách. Protože jde o silné endogenní organické kyseliny (pK po řadě 3,8 a 4,8), vyvolávají metabolickou acidózu. Kyselina 3-ketomáselná kromě toho spontánně dekarboxyluje, takže vzniká těkavý aceton, který je cítit z dechu nemocných.
V klinické praxi je nutno mít na paměti, že anionty obou uvedených silných organických kyselin jsou – coby reziduání anionty – tzv. potenciálními hydrogenuhličitany. Přívlastek „potenciální“ je odvozen z faktu, že oba reziduální anionty se při léčbě (aerobně) metabolizují za spotřeby H+ iontů. Jako čistý zisk této reakce vycházejí hydrogenuhličitany (obr. 6). Tato skutečnost je nesmírně závažná v terapeutické fázi. Rychlá přeměna potenciálních hydrogenuhličitanů na hydrogenuhličitany vyvolaná terapií ohrožuje pacienta smrtícím zvratem acidózy do alkalózy. Proto je při léčbě zapotřebí náležité opatrnosti.
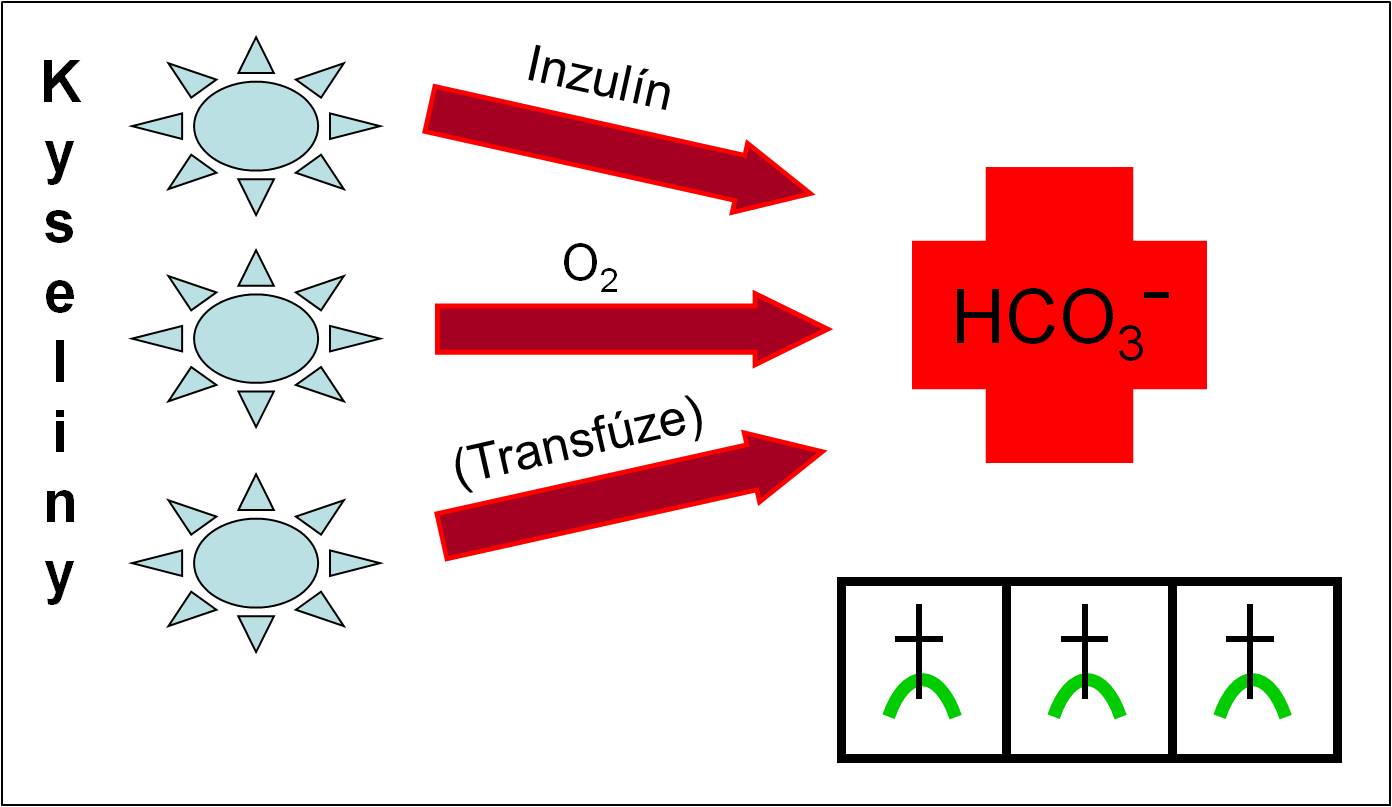
Obr. 6. Schéma životu nebezpečné přeměny potenciálních hydrogenuhličitanů na hydrogenuhličitany vyvolané terapeutickými zásahy. Schéma je možno zhlédnout i v animované podobě.
Sekundární procesy při ketoacidóze
Sekundární procesy se při pohybu koncentrací iontů H+ v plazmě rozvíjejí ve dvou rovinách – jednak v rovině chemické a jednak v rovině fyziologické. Obě základní komponenty acidobazické soustavy – komponenta respirační a komponenta nerespirační (metabolická) – při tom neustále zůstávají propojeny soustavou chemických pufrů tělních tekutin.
Sekundární obranná nárazníková reakce na nahromadění silných ketokyselin (H+) v extracelulární tekutině přímo zahrnuje jak hydrogenuhličitanové, tak nehydrogenuhličitanové pufry (z nich hlavně proteiny): 2H++ HCO3‾ + Prot‾ → H2O + CO2 + ProtH. Je nutno mít na paměti, že nejde o přímé (absolutní, jako např. při průjmu), nýbrž o nepřímé ztráty hydrogenuhličitanů.
Ketoacidóza se tak jako každá jiná abnormální acidobazická změna v metabolické složce zobrazí jako patologická hodnota výchylky bazí (v tomto případě deficit, tedy – BE).
Schodek aniontů, AG, definovaný jako AG (mmol/l) = ([Na+] + [K+]) – ([Cl‾] + [HCO3‾]), na jehož hodnotě se také podílejí koncentrace reziduálních aniontů, se nahromaděnými ketokyselinami zvětšuje. Vzniká normochlorémická acidóza provázená zvýšeným schodkem aniontů na úkor hydrogenuhličitanů.
V úrovni fyziologické má odpověď na primární změnu acidobazické rovnováhy podobu kompenzačních anebo korekčních reakcí zprostředkovaných specifickými orgány anebo orgánovými systémy regulace acidobazické rovnováhy, hlavně plícemi, ledvinami a játry.
Respirační adaptace na diabetickou ketoacidózu
Acidóza má stimulační účinky na dechové centrum. Odpověď dechového centra se rozvíjí do plné síly v průběhu 12 – 24 hodin, za asi 6 – 12 hodin nabývá asi 70 % maximální intenzity. Stejná doba je potřebná k odeznění odpovědi po úpravě poruchy (obr. 7).
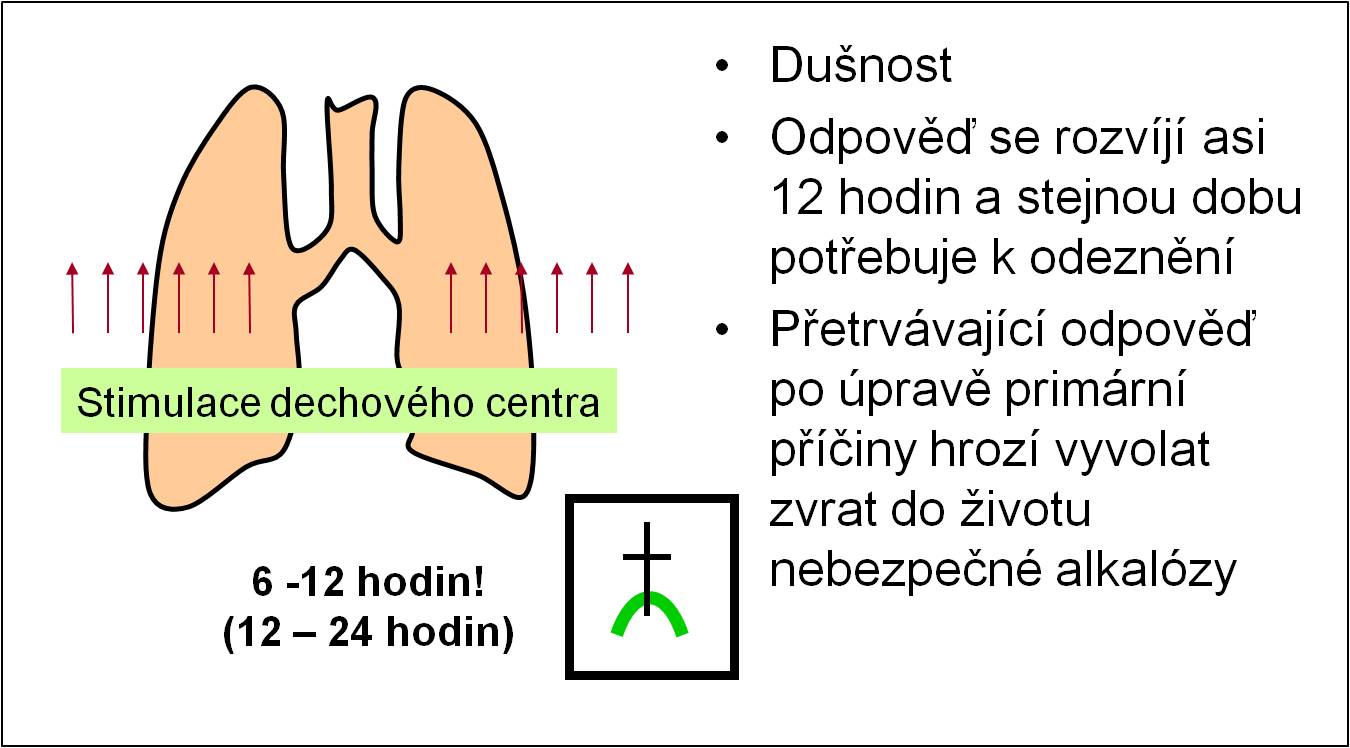
Obr. 7. Shrnutí kritických účinků acidózy na respirační systém. Schéma je možno zhlédnout i v animované podobě.
Respirační kompenzace vede k přeměně akutní prosté ketoacidózy na smíšenou (kombinovanou). Po rozvinutí respirační kompenzační odpovědi do maxima se pacient ocitá v pásu ustálené metabolické acidózy (obr. 8). Respirační odpověď zpravidla provází Kussmaulovo dýchání.
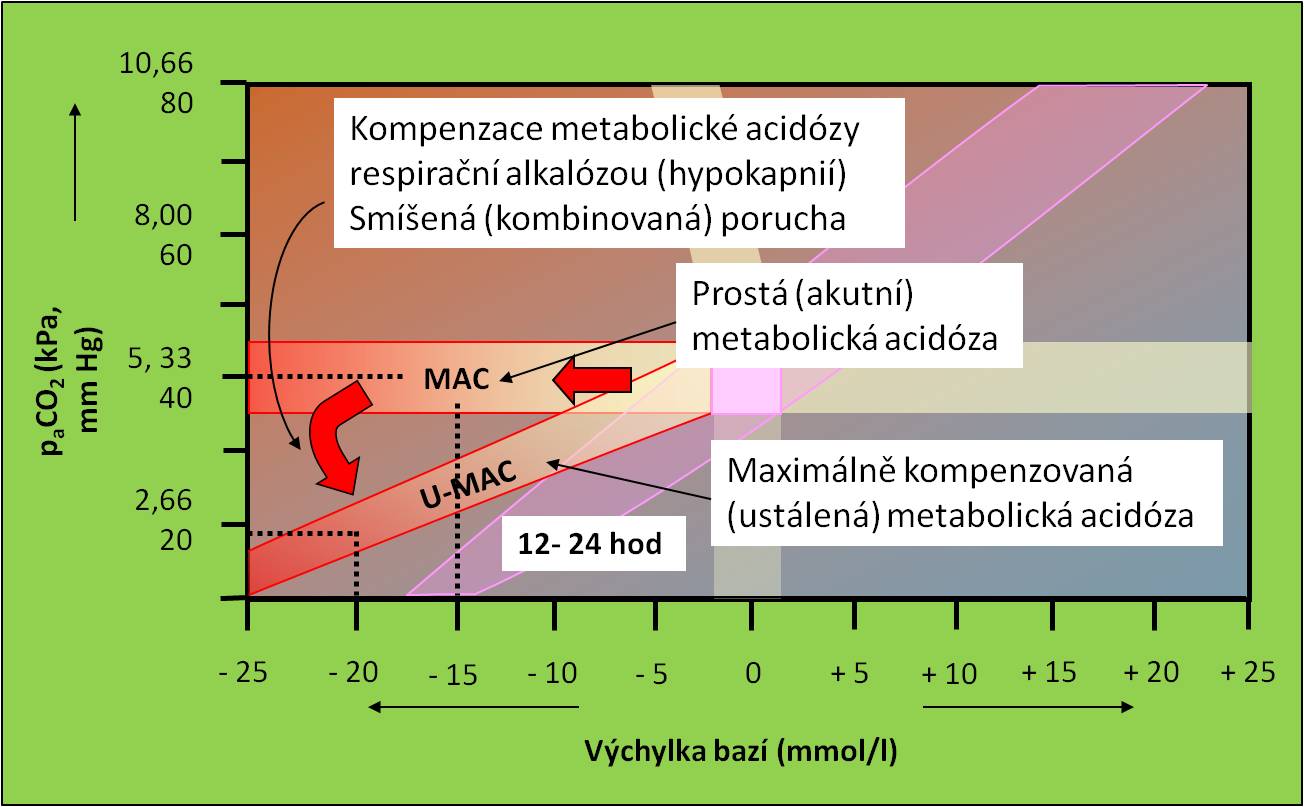
Obr. 8. Křížový graf acidobazické rovnováhy. Postup rozvoje diabetické ketoacidózy od prosté (akutní) poruchy přes smíšenou až k ustálené poruše. Schéma je možno zhlédnout i v animované podobě.
Renální adaptace na diabetickou ketoacidózu
Kompenzační odpověď ledvin na acidózu zahrnuje jak zvýšené vylučování titrovatelné kyseliny, tak zvýšenou exkreci NH4+. Množství hydrogenuhličitanů, vytvořených v ledvinách v procesu acidifikace moči a vrácených do organismu, je rovno součtu titrovatelné acidity a množství kationtu NH4+ v definitivní moči. Protože vylučování titrovatelné kyseliny se zvyšuje rychle, ale málo, spočívá mechanismus adaptace především v rozvoji sekrece NH4+ (obr. 9). Vzestup sekrece NH4+ při acidóze může dosahovat velikosti dvou řádů (zatímco při alkalóze klesá vylučování NH4+ prakticky na nulu).
Odpověď ledvin na acidobazickou poruchu se rozvíjí do plné síly v průběhu 5 – 7 dnů, za asi 3 – 5 dnů nabývá své 70 % maximální intenzity. Stejná doba je ovšem potřebná k odeznění odpovědi po úpravě poruchy.
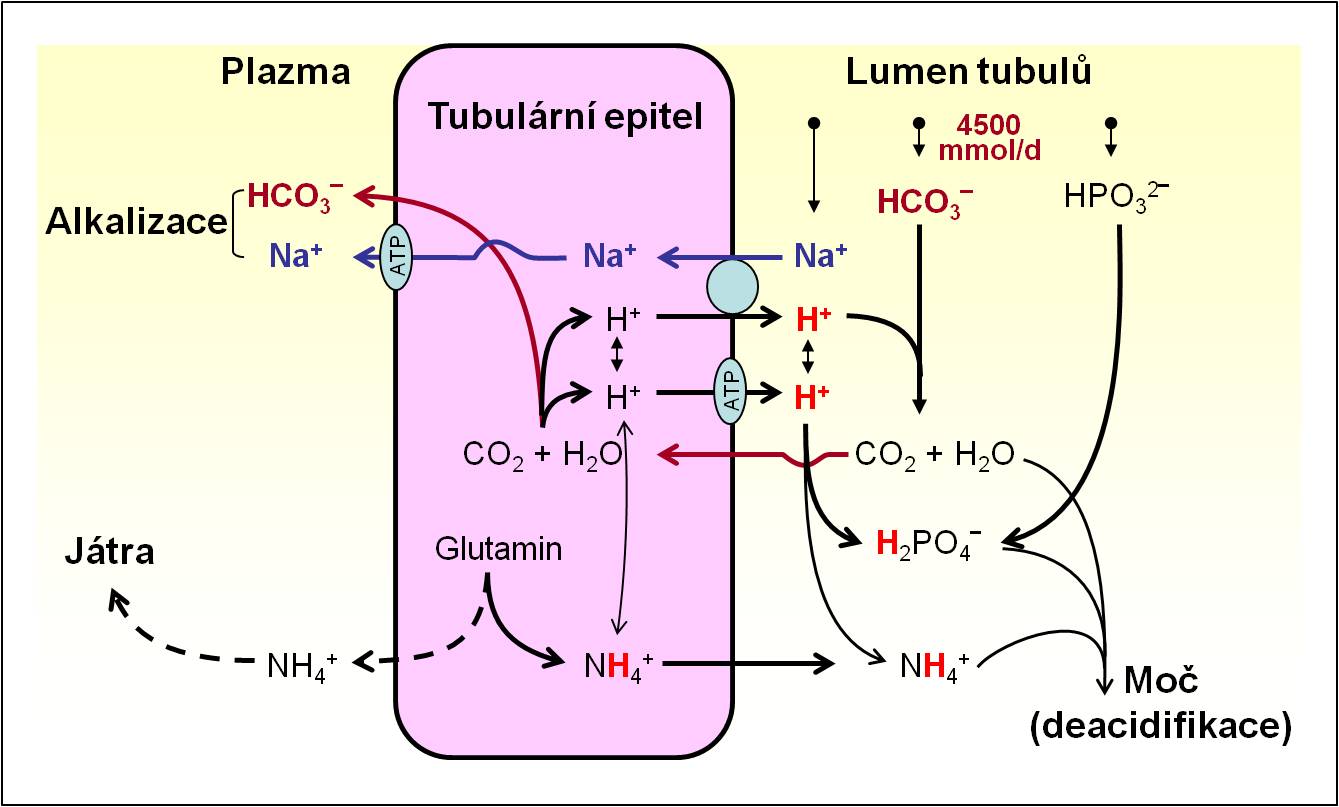
Obr. 9. Schéma renální adaptace na acidobazické poruchy. Schéma je možno zhlédnout i v animované podobě.
Přetrvávající ledvinové děje po odstranění primární příčiny acidózy mohou vést ke zvratu do životu nebezpečné alkalózy. Uvedené nebezpečí zvratu je v prvních nejméně 6 – 12 hodinách zesíleno přetrvávajícími respiračními kompenzačními ději.
Další klinicky významné účinky diabetické ketoacidózy
Účinky ketoacidózy na centrální nervový systém
Objevují se následující známky toxického poškození neuronů H+ ionty (obr. 10):
- Neklid, třes, poruchy reflexů;
- Psychické změny;
- Kóma.
Acidóza také má vazodilatační účinky na arterioly v centrálním nervovém systému. Tyto účinky jsou u respirační acidózy ještě zesíleny hyperkapnií, jejíž přítomnost přispívá k vysvětlení dalších příznaků ze strany centrálního nervového systému:
- Bolesti hlavy;
- Edém papily.

Obr. 10. Shrnutí kritických účinků acidózy na centrální nervový systém.
Finální stádium acidózy
Finálním stádiem diabetické acidózy je acidotické koma (obr. 11). Mají na něm hlavní podíl denaturační účinky H+ iontů na proteiny neuronů v centrálním nervovém systému. Často k němu přispívají další faktory, jako jsou dehydratace a selhání oběhu.

Obr. 11. Finální stádium acidózy. Schéma je možno zhlédnout i v animované podobě.
Účinky acidózy na kardiovaskulární systém
- Acidóza má negativně ino- i chronotropní účinky na myokard. Tyto účinky jsou zčásti kompenzovány vyplavením katecholaminů; u pacientů s β-blokátory však jsou účinky katecholaminů potlačeny a vliv acidózy na myokard se rozvíjí naplno (obr. 12).
- Acidóza spolu s katecholaminy zvyšuje pohotovost myokardu k arytmiím. Je dobře známo, že korekce acidózy velmi usnadňuje úpravu arytmií včetně defibrilace (obr. 12).
- Acidóza má vazokonstrikční efekt na periferní kapacitní vény. Proto při infúzní terapii zároveň zvýšeně hrozí objemové přetížení a následný edém plic. Zároveň má vazodilatační účinek na většinu systémových arteriol (obr. 12).
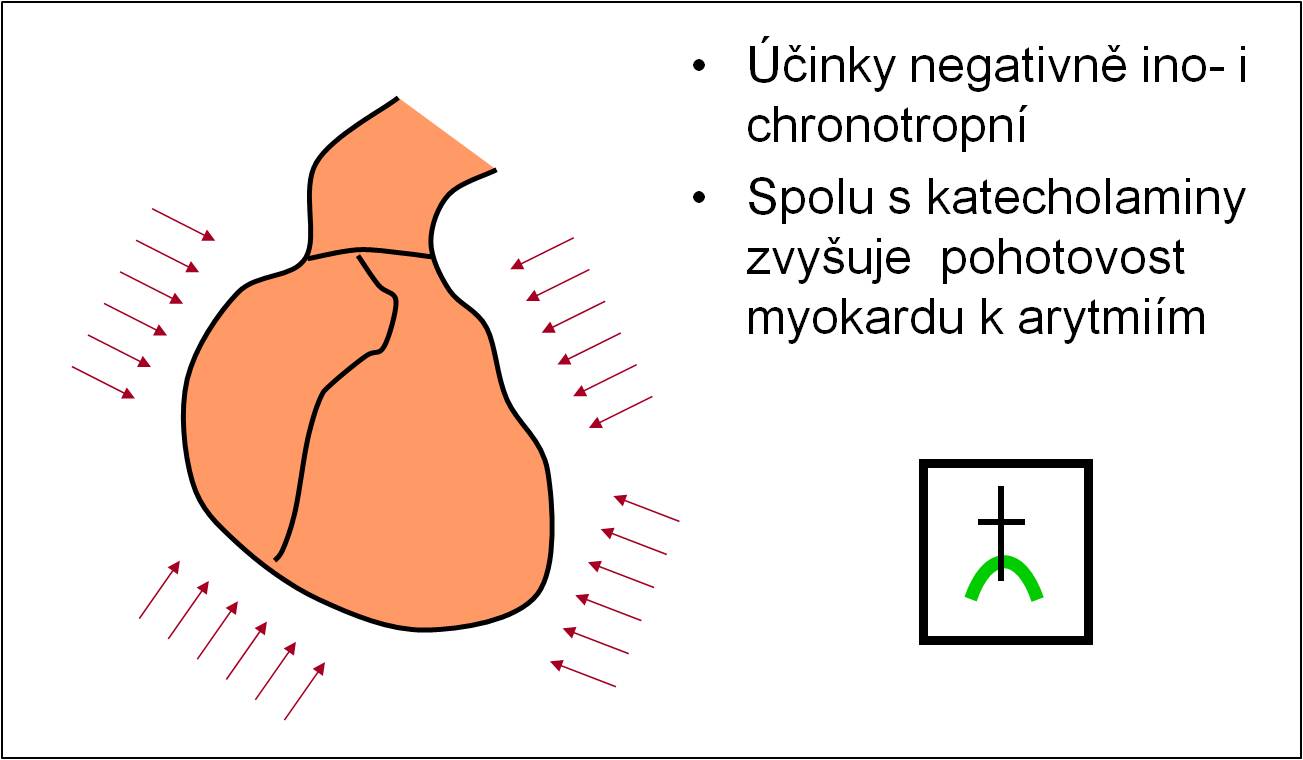
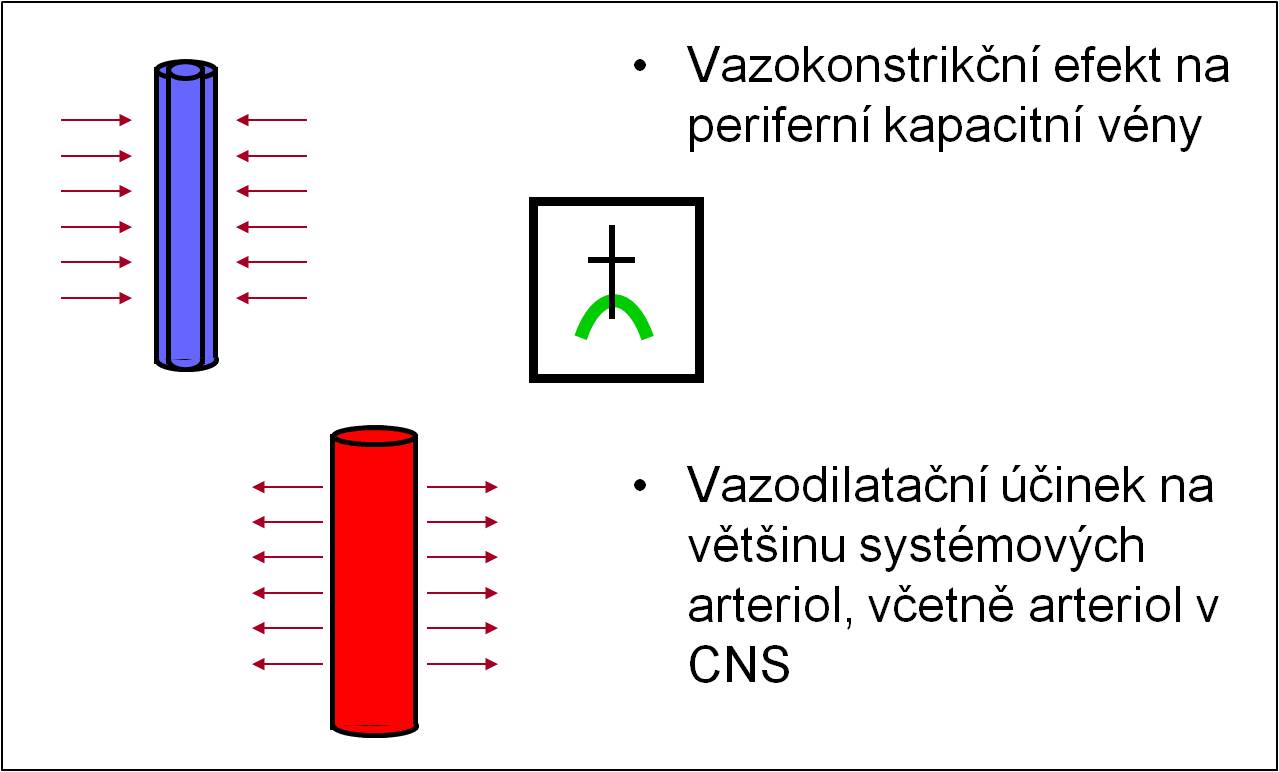
Obr. 12. Shrnutí kritických účinků acidózy na srdce a na cévy. Schéma je možno zhlédnout v animované podobě.
Účinky acidózy na hospodaření s ionty K+
Zvýšení koncentrace H+ iontů v extracelulární tekutině má za následek intenzívní vstup iontů H+ do buněk. Intracelulární ionty K+ se naproti tomu přesouvají do extracelulárního prostředí. Normální hodnoty sérových koncentrací K+ se proto při acidémii redefinují, jak ukazuje tabulka 1.
| pH | Normální hodnoty K+ (mmol/l) |
| 6,8 | 6,5 – 8,0 |
| 7,1 | 5,5 – 6,5 |
| 7,3 | 5,2 |
| 7,4 | 4,5 |
| 7,7 | 3,5 |
Tabulka 1. Redefinvané normální hodnoty koncentrace K+ v séru při acidémii.
Rozvíjející se hyperkalémie stimuluje sekreci aldosteronu. Proto po dobu acidózy dochází k vysokým ztrátám K+ iontů močí (obr. 13). V těle se tak rozvíjí těžký deficit K+, a to přes zdánlivou přítomnost hyperkalémie (tabulka 1). Při terapeutické úpravě acidózy pak dochází k překotnému návratu K+ iontů zpět do buněk (viz níže, obr. 16), ubývá iontů K+ v plazmě, a pacient je ohrožen těžkou hypokalémií, která může být smrtící. Proto musí terapii acidóz nezbytně provázet úhrada ztrát K+ iontů jako součást infúzí.
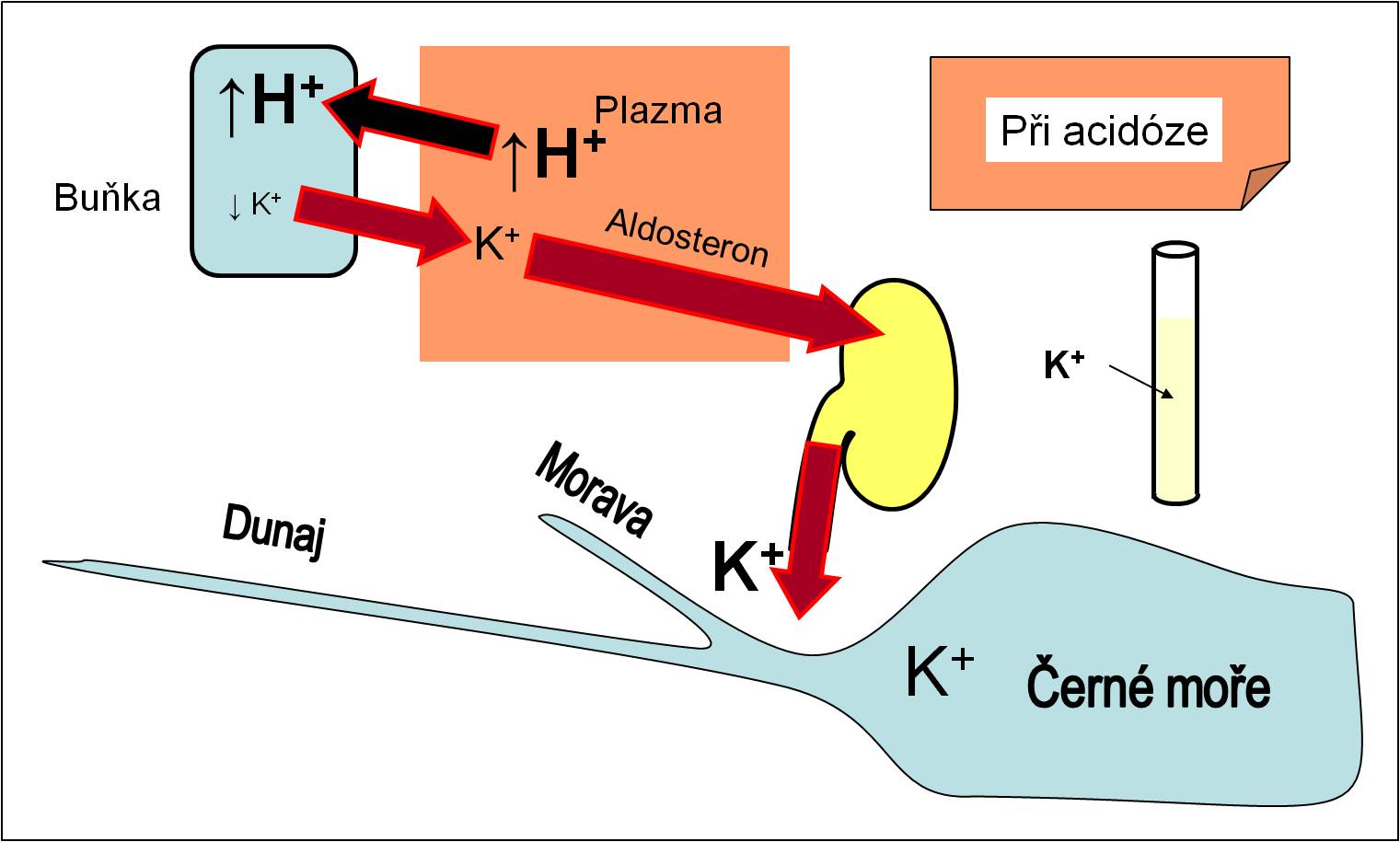
Obr. 13. Účinky acidózy na bilanci K+ iontů. V průběhu acidózy se vyvíjí těžká intracelulární deplece iontů K+ a deficit K+ iontů v organismu. Při nesprávě vedené terapii pak hrozí smrtelná hypokalémie. Schéma je možno zhlédnout i v animované podobě – jednak stav před acidózou, jednak procesy při acidóze.
Zásady terapie diabetické hyperglykemické ketoacidózy
1. Rehydratace
Jak už bylo ouvedeno, na prvním místě dbáme na funkce oběhového systému, tedy na rehydrataci (spolu s dostatečnou oxygenací).
2. Prevence mozkového edému
Pro prevenci náhlého poklesu osmolality plazmy používáme volumové expandéry (nelze provádět glukózou!).
3. Prevence zvratů jedné acidobazické poruchy v opačnou
Z pravidla kauzálního přístupu při léčbě poruch acidobazické rovnováhy vyplývá, že pacienta zásadně „netitrujeme“. To v plné míře platí u diabetického ketoacidotickéhostavu. Korekční terapii acidózy hydrogenuhličitany se nasazuje jen tam, kde skutečně jde o deficit HCO3‾. Ketokyseliny samy jsou „potenciálními hydrogenuhličitany“. V průběhu léčby inzulínem hydrogenuhličitany vznikají z aniontů ketokyselin v molárním poměru 1:1. Proto je podání hydrogenuhličitanů při léčbě ketoacidózy přísně kontraindikováno. Nedodržení tohoto pravidla zvyšuje fatální nebezpečí akutního zvratu acidózy v alkalózu (obr. 14).
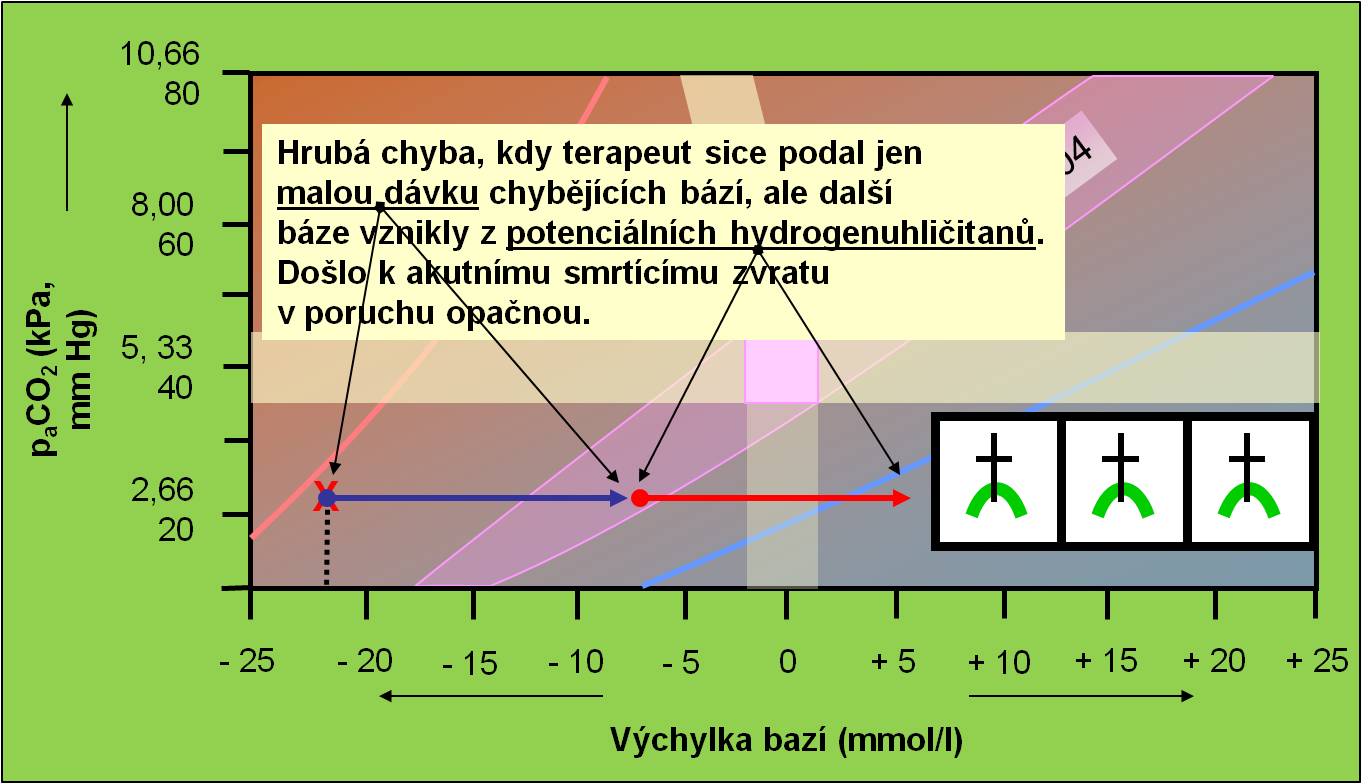
Obr. 14. Ilustrace náhlého zvratu z acidózy do alkalózy vyvolaného nesprávným podáním dávky hydrogenuhličitanů, která se sečetla s anionty vzniklými při metabolizaci potenciálních hydrogenuhličitanů. Schéma je možno zhlédnout i v animované podobě.
Další nebezpečí akutního zvratu acidózy do alkalózy vyplývá z podcenění účinků rozvinutých sekundárních kompenzačních a korekčních mechanismů v těle. Ustálená sekundární respirační odezva, provázející primární poruchu, potřebuje po odstranění primární příčiny acidózy minimálně 6 – 12 hodin, aby opět odezněla. Po tuto dobu může přetrvávající hyperventilace způsobit zvrat do životu nebezpečné alkalózy. Sekundární metabolická korekční odpověď, reprezentovaná hlavně ledvinami a játry, potřebuje ještě výrazně delší čas, přinejmenším 3 – 5 dnů, aby vyhasla. Po tuto dobu je nutno zajistit, aby hrozící zvraty neohrozily život pacienta (obr. 15).
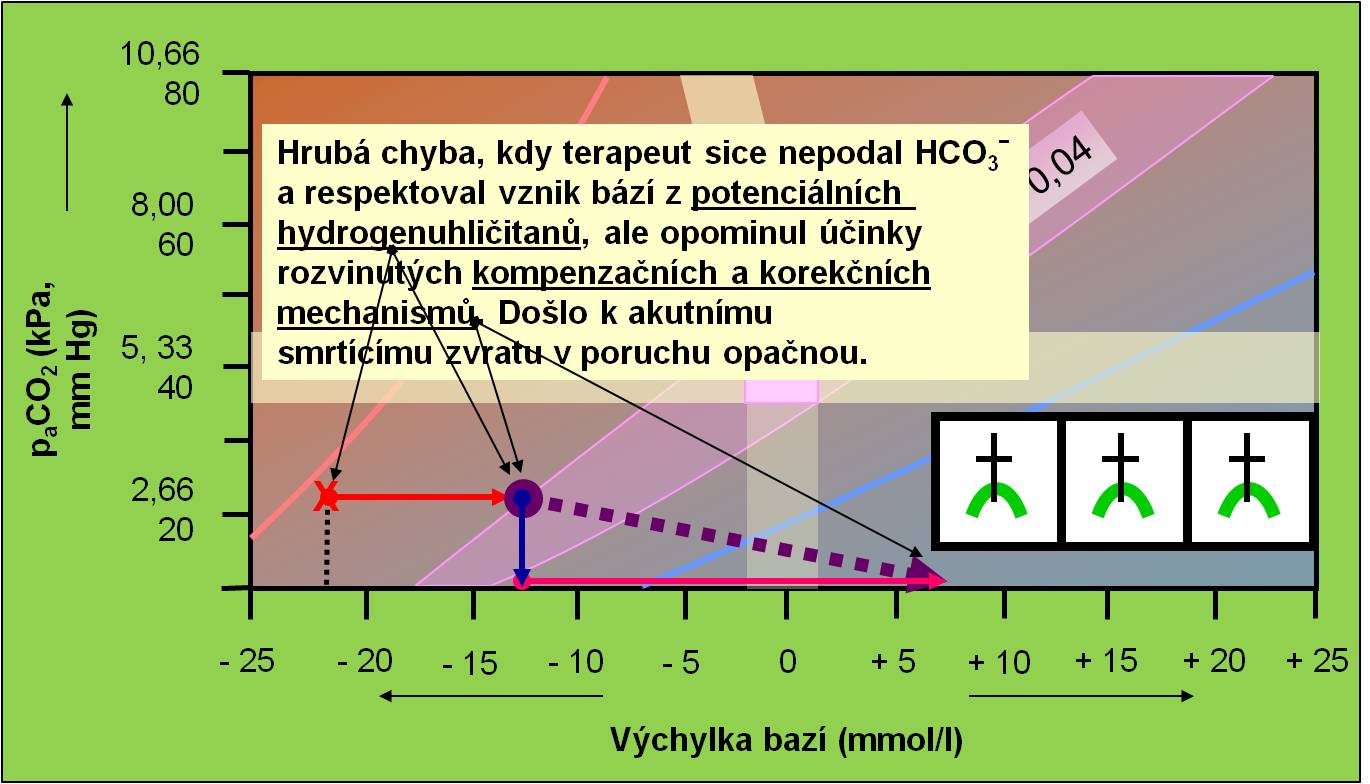
Obr. 15. Ilustrace náhlého zvratu z acidózy do alkalózy, kdy lékař sice nepodal hydrogenuhličitany (SPRÁVNĚ!) a vzal v úvahu metabolizaci potenciálních hydrogenuhličitanů (SPRÁVNĚ!), ale opomnněl přetrvávající účinky rozvinutých kompenzačních a korekčních mechanismů (respirační systém: 6 – 12 hodin, metabolický systém: 3 – 5 dnů!). Schéma je možno zhlédnout i v animované podobě.
4. Prevence iontových zvratů
Dalším urgentním rizikem při terapii acidózy je hrozící hypokalémie z náhlého přesunu K+ iontů z krevní plazmy do cytoplazmy buněk po nasazení terapie inzulínem a navození anabolismu. Vzhledem k deficitu K+ iontů, který se v průběhu acidózy rozvíjí, nejsou zásoby K+ iontů v těle dostatečné pro nasycení intracelulárních prostor. Vzniklá hypokalémie může být smrtící (obr. 16). Prevencí může být jen dostatečný přívod exogenního kalia infúzí.
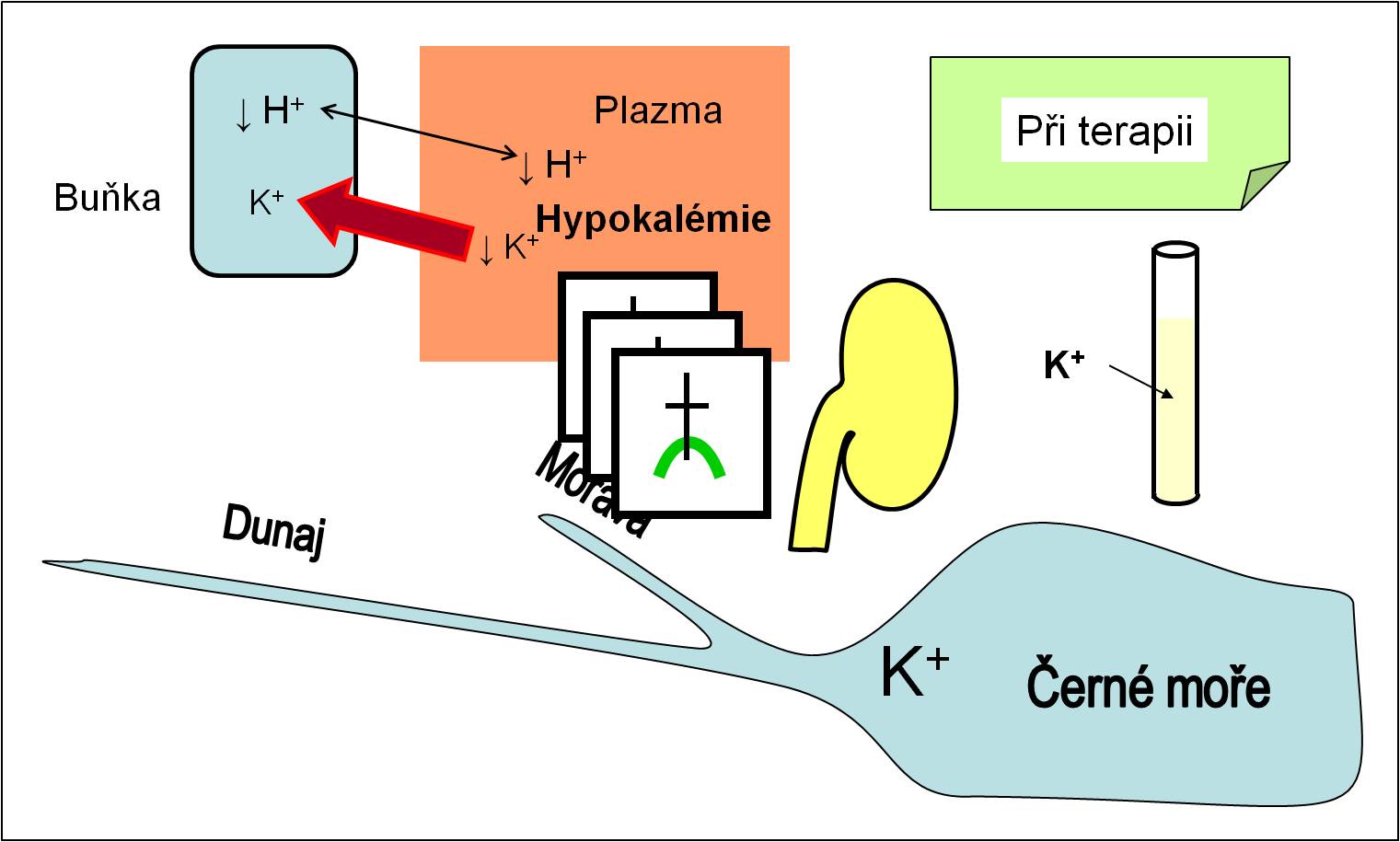
Obr. 16. Účinky léčby acidózy na plazmatickou koncentraci K+ iontů. Ionty K+ se v průběhu léčby transportují zpátky do buněk. Při terapii acidóz tak hrozí smrtící hypokalémie. Schéma je možno zhlédnout i v animované podobě.
Klinický případ diabetické ketoacidózy je popsán v kazuistikce uvedené na této adrese.
Diabetické laktacidotické kóma
Laktacidotické kóma se nevyskytuje často. Je průvodním vedlejším (nežádoucím) jevem při léčbě perorálními antidiabetiky typu biguanidů. Riziko vzniku laktacidotického kómatu se zvyšuje při současném onemocněních levin, jater anebo srdce. Přitom glykémie je jen mírně zvýšená, a hyperosmolarita proto není v popředí klinického obrazu anebo komplikací.
Diabetická laktacidotická kóma se projevuje nástupem komatózního stavu (slabost, únava, spavost, netečnost) a sklonem k arteriální hypotenzi. Laktát bývá dramaticky zvýšen (i nad 10 mmol/l), je přítomna krajně těžká acidóza (pH 6,8). Jde o jednu z absolutních indikací k terapii dialýzou. I přes intenzivní terapeutický zásah však tato komplikace často bývá smrtelnái.
Hypoglykemické diabetické kóma
Hypoglykémie je stav s koncentrací krevní glukózy pod 3,4 mmol/l.
Nejčastější příčinou diabetického hypoglykemického kómatu je nepřiměřeně absolutně nebo relativně vysoká dávka inzulínu a vynechaná anebo snížená dodávka stravy po dávce inzulínu. Jindy je to nadměrná tělesná námaha, alkohol atd. Těžká iatrogenní hypoglykémie se může rozvinout i na nemocničním oddělení, pokud personál nemocnému aplikuje předepsanou dávku inzulínu, ale už nedohlédne na – nebo se nepostará o – to, aby pacient dostal – a také požil – příslušnou dávku stravy.
Při hypoglykémii je možná záměna s cévní mozkovou příhodou (fatální „nová ataka“ CMP na neurologickém oddělení při právě uvedeném zanedbání péče personálem) anebo – běžněji v denním životě – s opilostí, epilepsií anebo jiným záchvatem.
Neuroglykopenické příznaky zahrnují iritovanost, zmatenost anebo poruchy řeči, vidění, pozornosti, chování, křeče,pak útlum a kóma. Příznaky vyplývající z alarmové aktivace sympatického systému se manifestují tachykardií, bledostí, zpocenou chladnou kůží, třesem, úzkostí, hladem atd. Kůže však někdy paradoxně může být červená z periferní vazodilatace. Přehled podává tabulka 2.
Nástup hypoglykemického kómatu je u osob douhodobě trpících diabetem usnadněn opožděnou signalizací hypoglykémie způsobenou diabetickou autonomní neuropatií.
| Glykémie (mmol/l) | Symptomy |
| < 3,8 | Zvýšení sekrece glukagonu, katecholaminů, glukokortikoidů |
| < 3,2 | Neklid, intenzivní pocit hladu, poruchy pozornosti, pocení, bušení srdce, záchvěvy či třes (rukou), úzkost, poruchy psychomotorických funkcí |
| < 2,8 | Poruchy kognitivních funkcí |
| < 2,0 | Poruchy vědomí, křeče, kóma |
Tabulka 2. Příznaky hypoglykémie.
Klinické symptomy a diagnóza diabetu
Diabetes se často vůbec poprvé manifestuje některou z akutních hyperosmolárních komplikací popsaných výše. Jindy pacienti s diabetem přicházejí k vyšetření pro některý ze symptomů hyperglykémie.
Hlavní klinické symptomy hyperglykémie jsou:
- Polyurie;
- Polydipsie;
- Zvýšená chuť k jídlu a zvýšený příjem potravy;
- Ztráta hmotnosti;
- Poruchy růstu;
- Náchylnost k infekcím, zejména kvasinkovým a plísňovým, špatně se hojící poranění, nehojící se bércové vředy;
- Poruchy zraku;
- Poruchy krevního zásobení orgánů a končetin.
Chronické komplikace diabetu strmě narůstají při koncentracích glukózy v plazmě nad 6,9 mmol/l (obr. 1).
Základní laboratorní vyšetření nemocných diabetem
Níže jde o stručný výtah doporučení podaného expertní skupinou České společností klinické biochemie ČLS JEP. Originální materiál je dostupný na interntové adrese.
Stanovení glukózy v plazmě anebo v séru
Stanovení glukózy v plazmě anebo v séru se provádí ve vzorcích krve odebrané nalačno, obvykle po hladovění přes noc, nejméně však po 8 hodinách (FPG, fasting plasma glucose), s vyloučením fyzické námahy a kouření. Výsledek stanovení má pro diagnózu diabetu základní význam. Meze jsou uvedeny v tabulce 3. Dva instruktážní videoklipy zachycující měření glykémie automatickým laboratorním analyzátorem a příručním glukometrem jsou dostupné na této adrese.
| FPG (mmo/l) | Výklad |
| 3,4 – 5,6 | Normální hodnoty glukózy v plazmě anebo v séru |
| 5,6 – 6,9 | Zvýšená koncentrace glukózy nalačno, svědčí pro poruchu glukózové tolerance (IFG, impaired fasting plasma glucose, nebo IGT, impaired glucose tolerance), mezistav mezi normální homeostází glukózy a diabetem; prediabetes.Nutno ověřit opakovanými vyšetřeními. |
| ≥ 7,0 | Svědčí pro klinickou dg. diabetes mellitus.Nutno potvrdit opakovanými vyšetřeními. |
| ≥ 11,1 | Při přítomnosti dalších klinických symptomů svědčí pro klinickou dg. diabetes mellitus. Nesmí jít např. o hyperglykémii vyvolanou akutním stresem – po úrazu apod. – jde o častý omyl zejména chirurgických (traumatologických) oborů. |
Tabulka 3. Meze hodnocení koncentrací glukózy v plazmě anebo v séru.
Stavy IGF anebo IGT samy o sobě nejsou klinickými jednotkami (vyjma situace u gestačního diabetu – viz níže). Jsou známkou zvýšeného rizika vzniku diabetu. Navíc, přísně vzato, je namístě dělat rozdíl mezi IGF a IGT; řada pacientů s IGT může mít po většinu času euglykemické koncentrace glukózy, které překračují normy pouze po zátěži. Zejména diabetes 2. typu se však zpravidla vyvíjí dlouho, plíživě a jeho smyptomy nebývají nápadné, takže často bývá rozpoznán poměrně pozdě, až v pokročilém stádiu orgánových změn.
Při diagnostických rozpacích (IFG) je indikován orální glukózový toleranční test (oGTT). Zkouška oGTT je rovněž indikována u těhotných majících zvýšené riziko diabetu (pak se provádí co nejdříve v prvním trimestru gravidity) a u každé těhotné ve 24 – 28. týdnu gravidity, pokud nemá nízké riziko gestačního diabetu. Hodnotí se koncentrace glukózy v plazmě žilní krve nalačno a za 2 hodiny po p.o. podání 75 g glukózy. Meze jsou uvedeny v tabulkách 4 a 5.
| Glukóza (mmo/l) | Výklad |
| < 7,8 | Nesvědčí pro diabetes |
| 7,8 – 11 | Porušená tolerance glukózy |
| ≥ 11,1 | Diabetes |
Tabulka 4. Meze hodnocení oGTT. Koncentrace glukózy v žilní plazmě za 2 hodiny po podání glukózy.
| Glukóza (mmo/l) | Výklad |
| FGP ≥ 5,6 | Gestační diabetes |
| Za 2 hodiny po podání ≥ 7,7 | Gestační diabetes |
Tabulka 5. Meze hodnocení oGTT u těhotných. Koncentrace glukózy v žilní plazmě nalačno a za 2 hodiny po podání glukózy.
Kritéria nízkého rizika gestačního diabetu, jež všechna musí být splněna, jsou následující:
- Těhotná žena mladší 25 let;
- Má normální tělesnou hmotnost;
- Nemá výskyt diabetes mellitus v rodinné anamnéze u nejbližších příbuzných (příbuzní 1. stupně);
- Je běloška anebo není příslušnicí etnické skupiny nebo rasy se zvýšeným výskytem diabetu.
Měření koncentrace glukózy v kapilární krvi osobními glukometry
Osobní glukometry stanovují koncentraci glukózy v kapilární krvi. Využívají je pacienti s diabetem léčení inzulínem (diabetes 1. typu) a někteří pacienti trpící diabetem 2. typu. Je dostupný příklad měření osobním glukometrem a příkady možností použití kontinuálního měření glykémie a inzulínové pumpy.Výsledky měření glukometry vyráběnými různými výrobci mezi sebou příliš nesouhlasí.
Plazma kapilární a žilní krve má stejnou koncentraci glukózy. Po zátěži glukózou (po jídle, po podání glukózy) je koncentrace glukózy v plazmě kapilární krve asi o 25 % vyšší.
Koncentrace ketolátek
Koncentrace ketolátek u nemocných s diabetem se stanovuje vždy při výskytu symptomů ketoacidózy anebo při glykémii vyšší než 16,7 mmol/l. Význam má stanovení ketolátek v krvi. Rutinní metody měření však postihují pouze kyselinu 3-ketomáselnou (neboli acetoctovou) a aceton (tedy ketony). Kyselina 3-hydroxymáselná, která je redukovaným ekvivalentem kyseliny 3-ketomáselné, se s ní za aerobních podmínek v tělních tekutinách vyskytuje v ekvimolárním množství. Při tkáňové hypoxii provázející diabetickou ketoacidózu se však rovnováha posunuje na stranu kyseliny 3-hydroxymáselné. Rutinně stanovované koncentrace ketolátek tedy mohou významně podhodnoceny.
Glykovaný hemoglobin Hb1c
Normální hodnoty glykovaného hemoglobinu Hb1c jsou 20 – 42 mmol/mol. Hodnoty mezi 43 – 53 mmol/l svědčí pro kompenzovaný diabetes.
Koncentrace albuminu v moči, proteinurie
Zvýšené koncentrace albuminu v moči obecně indikují endotelovou dysfunkci a jsou včasným ukazatelem kardiovaskulárního rizika. U diabetu jde o významný indikátor vývoje diabetické nefropatie. Závěry se dělají až na základě opakovaných měření.
První známkou diabetické nefropatie, kdy vylučovaná množství albuminu ještě nejsou postižitelná rutinními metodami testování moči, je tzv. mikroalbuminurie (množství albuminu 20 – 200 µg/minutu anebo 30 – 300 mg/24 hodin). Zjišťuje se imunochemicky anebo vyšetřením pomocí HPLC. Koncentraci albuminu lze vztáhnout ke koncentraci kreatininu (ACR, albumin-creatinin ratio) – hodnoty mikroalbuminurie jsou mezi 3,5 – 30 (g albuminu/mol kreatininu). Nad tyto meze už jde o proteinurii.
Stanovení C-peptidu anebo inzulínu
Koncentraci C-peptidu je vhodné stanovit u pacientů trpících diabetem 2. typu, pokud je podezření na selhání sekrece inzulínu anebo se rozhoduje o terapii inzulínem. Stanovení inzulínu přispívá k posouzení inzulínové rezistence při zřetelném vyjádřené klinické symptomatologii (např. u syndromu polycystických ovarií). Mimoto patří k základním vyšetřením při podezření na inzulinom (nesidiom).
Na aktuální koncentraci C-peptidu anebo inzulínu má vliv fyzická zátěž a kouření, z léků pak užívání vitamínu B7 (vitamín H, biotin).
Imunologické vyšetření
Imunologické vyšetření doplňuje vyšetření pacientů při podezření na autoimunitní onemocnění. Diabetes se nezřídka sdružuje s autoimunitními onemocněními (sdružené autoimunity) jiných endokrinních žlaz anebo orgánů včetně perniciózní anémie, vitiliga, céliakie (němá forma céliakie) atd.
- Až 90 % dětských pacientů trpících diabetem 1. typu má protilátky proti inzulínu (IAA).
- Až u 80 % pacientů trpících diabetem 1. typu se vyskytují protilátky proti cytoplazmě ß-buněk pankreatu (ICA).
- Až u 80 % pacientů trpících diabetem 1. typu označovaným jako LADA (latentní autoimunitní diabetes dospělých) jsou přítomny protilátky proti dekarboxyláze kyseliny glutamové (anti-GAD); toto onemocnění je často zpočátku klasifikováno jako diabetes 2. typu.
- Asi u 50 % pacientů s diabetem 1. typu se vyskytují protilátky proti izoformám tyrosinfosfatázy (ostrůvkový antigen, islet antigen IA-2A, IA-2ßA).
Úskalí léčby diabetu
Somogyiho fenomén
Při léčbě inzulinem dochází k hypoglykémii v kteroukoliv denní dobu, mnohdy v době maximálního účinku inzulinu. Noční hypoglykémie ve spánku kolem 2. až 3. hodiny ranní jsou typické při aplikaci depotních inzulínů, zejména pokud se aplikují před večeří kolem 19. hodiny. Pokud pak následuje ranní posthypoglykemická hyperglykémie, nazývá se tento jev Somogyiho fenomén. Hyperglykémie je v tomto případě způsobena vysokými hladinami hormonů, které působí proti inzulínu, vyprovokovanými hypoglykémií.
Nutná opatření jsou následující:
- Sníst druhou večeři;
- Posunout dávku inzulinu na 22 – 23 hodin (účinek se posouvá k ranním hodinám, kdy fyziologicky narůstá rezistence vůči inzulínu).
Fenomén stmívání
Tzv. fenomén stmívání (dark phenomenon) je zvýšení glykémie v odpoledních hodinách u pacientů léčených inzulínem. Vyskytuje se zejména u nejmladších pacientů (děti asi do 6 let věku), kteří mají největší potřebu bazální hladiny inzulínu navečer. Hyperglykémie je v tomto případě způsobena zvýšením hormonů, které působí opačně než inzulín (zejména růstový hormnon anebo glukokortikoidy). S přibývajícím věkem pacientů se večerní bazální potřeba inzulínu obvykle snižuje, ale narůstá potřeba v ranních hodinách (komplikace se přesouvá k tzv. fenomén úsvitu).
Fenomén úsvitu
Tzv. fenomén úsvitu (dawn phenomenon) je zvýšení glykémie v ranních hodinách u pacientů léčených inzulínem. Opět je známkou nedostatečné kompenzace diabetu. Ani v tomto případě nedokáže běžné dávkování pokrýt bazální potřebu inzulínu při jeho zvýšené poptávce při pravidelném ranním zvýšení hormonů, které působí opačně než inzulín (mezi 4. – 7. hodinou). Lze mu předejít aplikací inzulínu kontinuální subkutánní infúzí inzulínovou pumpou.
Literatura a zdroje k dalšímu studiu
www zdroje:
The Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Summary of Revisions for the 2002. Diabetes Care January 2002 25:s5-s20. (staženoo 1. 5. 2012)
Fridecký B a spol.: Laboratorní diagnostika a sledování stavu diabetes mellitus. Klin. Biochem. Metab. 1 2006, 54-65. Staženo 12. 6. 2012.
Kalvodová a spol.: Doporučené postupy pro diagnostiku a léčbu diabetické retinopatie. Revize ke dni 14. 6. 2011. Staženo 12. 6. 2012.
Ambler Z: Diabetické neuropatie. Postgraduální medicína 3, 2004. Staženo 15. 7. 2012.
Rybka J: Diabetes mellitus – komplikace a přidružená onemocnění. Diagnostické a léčebné postupy. Praha, Grada, 2007. Staženo 15. 7. 2012.
Zpracoval: Jaroslav Veselý, Ústav patologické fyziologie LF UP v Olomouci

