

Hypoperfúze a ischémie mozku
Fyziologické základy
Ve shodě se základní rovnicí hemodynamiky (Q = P/R) je průtok krve mozkem (Qc) poměrem perfúzního tlaku v mozku (CPP, cerebral perfusion pressure) a cerebrovaskulární rezistence (Rc):
Qc = CPP/Rc.
Perfúzní tlak v mozku (CPP) je rozdílem mezi středním arteriálním tlakem (MAP) a intracerebráním tlakem (ICP): CPP = MAP – IPP (mm Hg).
Autoregulační mechanismy zajišťují stálou úroveň průtoku krve mozkovou tkání 50 ml/100 g tkáně/min. Autoregulace účinně pracuje v rozmezí arteriálního tlaku 60 – 150 mm Hg (obr. 1).

Obr. 1. Stálý průtok krve mozkem v rozmezí 60 – 150 mm Hg arteriálního tlaku zajišťovaný mpzkovou autoregulací.
Léčebně se dostatečné hodnoty CPP zajišťují zvýšením MAP anebo snížením ICP. Klinický příklad je uveden v kazuistice na adrese http://pfyziolklin.upol.cz/wp-content/uploads/2012/04/NitrolebHTKaz.pdf.
Patofyziologie ischémie mozku
Při snížení perfúze mozku pod asi 20 ml/100 g tkáně/min nastávají funkční reverzibilní změny (funkční práh). Manifestují se oploštěním EEG záznamu.
Při snížení mozkové perfúze pod asi 10 ml/100g tkáně/min (infarktový práh) na dobu přibližně 5 min a více dochází k nevratným změnám. Postup ischemických změn ilustruje spirála ischemické smrti (obr. 2).

Obr. 2. Spirála ischemické smrti neuronů.
| Čas | Popis děje |
| 8 s | Žádný kyslík |
| 10 – 12 s | Bezvědomí |
| 30 – 40 s | Vymizí odezva na EEG |
| 3 – 4 min | Spotřebována veškerá glukóza Začíná nekróza, smrt buněk |
| 8 – 9 min | Smrt mozku (Výjimka: Hypotermie) |
Tabulka 1. Spirála ischemické smrti neuronů v číslech.
Pokud jde o reakci vazomotorického centra na kritickou hypoperfúzi a hypoxii, nejprve se projevuje tím, že centrum po několik minut vysílá silné výboje navozující masívní aktivaci sympatiku (tzv. ischemická odpověď CNS). Toto poslední krizové vzepětí vazomotorické regulace ustává asi po 15 minutách, kdy už může aktivita centra zcela vymizet.
Etiologie lokalizované ischémie mozku (vyvolává ložiskové příznaky)
- Cévní spasmus;
- Arterioskleróza a trombóza;
- Embolizace;
- Poruchy vlastností krve;
- Jiné než aterosklerotické onemocnění cévní stěny.
Klasifikace cévních mozkových příhod
Za 85 % všech cévních mozkových příhod je odpovědná ischémie. Pouze 15 % všech mozkových příhod je způsobeno krvácením.

Obr. 3. Schéma klasifikace ischemických mozkových příhod.
Etiologie globální ischémie mozku (poškozuje celý mozek)
- Pokles perfúze mozku při selhání cirkulace;
- Hypoxémie při selhání respirace;
- Okluze karotid;
- Otrava CO.
Kazuistika: Difúzní edém mozku po celkové hypoxii
58letý pacient po transplantaci ledviny. Náhlá dušnost, edém plic, srdeční zástava, bezvědomí. Provedena úspěšná kardiopulmonální resuscitace, po které pacient zůstává v bezvědomí v kritickém stavu. S odstupem několika hodin provedeno CT vyšetření mozku (obr. 4a-d).




Obr. 4a-d. Na CT jsou patrné mapovité hypodenzní oblasti v celém mozku i mozečku, vyhlazení gyrifikace, komprese komorového systému a zánikem cisteren na bázi mozku. Jde o obraz difúzního edému mozku po celkové hypoxii (nedostatečném zásobení mozku kyslíkem). Hyperdenzní střední i přední mozečkové tepny („dense artery sign“) svědčí pro přítomnost trombů v nich a nepřímo i pro zástavu cirkulace krve mozkem.
Pacient po 4 dnech umírá.
Hypoperfúze a ischémie myokardu
Fyziologické základy
Srdeční sval má v přepočtu na jednotku hmotnosti jednu z nejvyšších spotřeb kyslíku mezi ostatními tkáněmi a orgány (tabulka 2). Zároveň má nejvyšší arteriovenózní diferenci koncentrací kyslíku (tabulka 3). Tyto vlastnosti jsou dány zvláštnostmi metabolismu v srdečním svalu, zejména vysokým podílem substrátů s vysokým respiračním kvocientem (mastných kyselin a ketolátek) na zajištění jeho energetických potřeb.
| Orgán | Spotřeba O2 (ml O2/min/100 g tkáně) |
| Mozek | 3 |
| Ledviny | 5 |
| Střevo v klidu | 2,5 |
| Kůže | 0,2 |
| Kosterní sval | |
| V klidu | 1 |
| Při námaze | 50 |
| Srdce | |
| Při zástavě | 2 |
| Klidový srdeční výdej | 8 |
| Těžká námaha | 70 |
Tabulka 2. Srovnání spotřeby kyslíku v orgánech.
| Orgán | A-V diference O2 (obj. % O2; ml O2/100 ml krve) |
| Mozek | 4 – 6 |
| Ledviny | 2 – 3 |
| Kůže | 1 – 2 |
| Střevo v klidu | 4 – 6 |
| Kosterní sval v klidu | 2 – 5 |
| Srdce (klidový srdeční výdej) | 10 – 12 |
Tabulka 3. Srovnání kyslíkové A-V diference v různých orgánech.
Koronární artérie se zanořují do myokardu z vnějšího povrchu a míři k vrstvám pod endokardem. Čím jsou jejich větve jemnější, tím jsou snáze stlačitelné. V izometrické fázi systoly v nich může krevní průtok klesat až k nule (obr. 5). Zásobení myokardu kyslíkem proto v průběhu cyklu značně kolísá. Z této skutečnosti vyplývá, jakou důležitost má pro funkci srdce přiměřené trvání fáze relaxace (diastoly). Jen tak může být zajištěna rovnováha mezi spotřebou a přívodem O2. Fyziologická rezerva průtoku krve koronárními cévami je asi 6násobná (3 – 9násobná) ve srovnání s klidovým stavem.
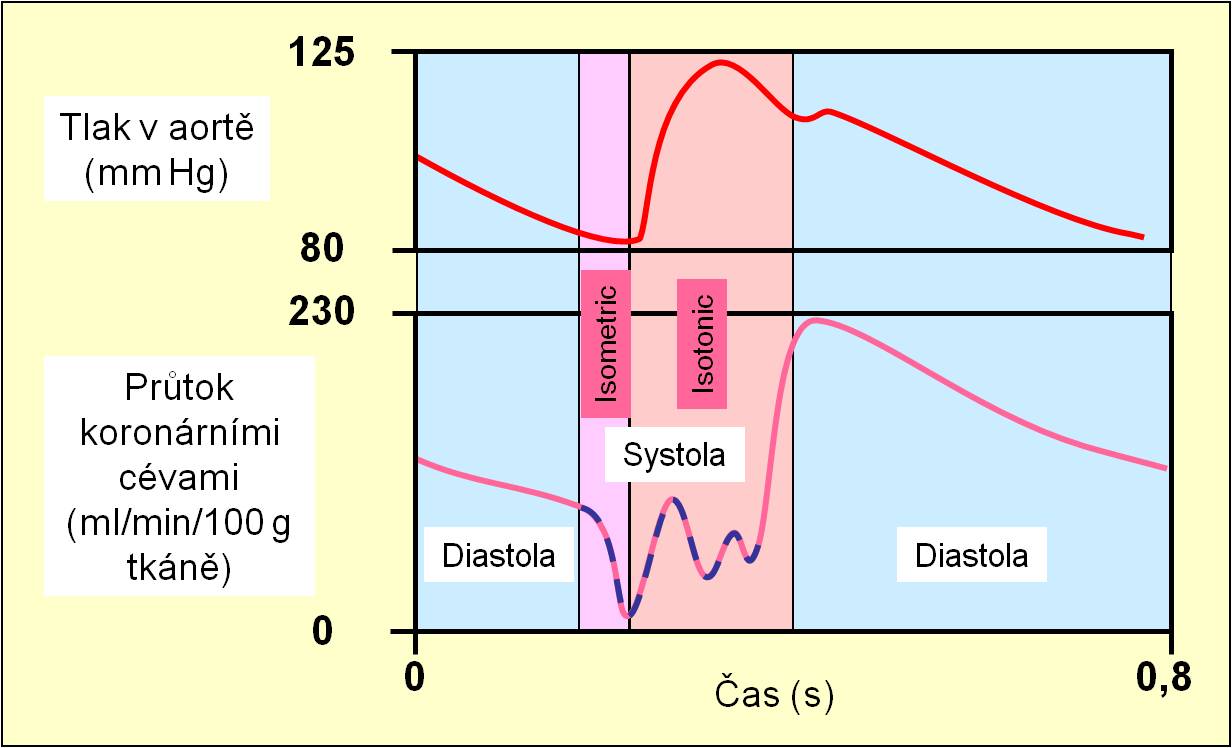
Obr. 5. Průtok krve myokardem v průběhu srdečního cyklu.
Patofyziologie ischémie myokardu
Hlavní nitrobuněčné změny po kritickém omezení přívodu krve a kyslíku jsou následující (obr. 6):
- Pokles ATP; nedostává se ATP pro udržení iontových gradientů na plazmatické membráně:
- Výstup K+ z buněk, vstup Na+ do cytoplazmy
- Je trvale přítomna částečná až úplná depolarizace srdečních svalových vláken v místě ischémie, je vysoká koncentrace iontů kalia extracelulárně v místě ischémie, iontové dysbalance zasahují i okolí ložiska; mají vliv i na vzrušivost a dráždivost vláken přímo nezasažených ischémií;
- Vstup Ca2+ do cytoplasmy
- Přetížení buněk kalciem;
- Výstup K+ z buněk, vstup Na+ do cytoplazmy
- Hromadění laktátu z anaerobních pochodů:
- Intra- a extracelulární acidóza v místě ischémie, šíří se i do okolí ložiska; má vliv i na vzrušivost a dráždivost vláken přímo nezasažených ischémií;
- Při dosažení kritické hranice ischémie dochází po 20 minutách k nevratnému rozpadu (nekróze) svalových vláken.
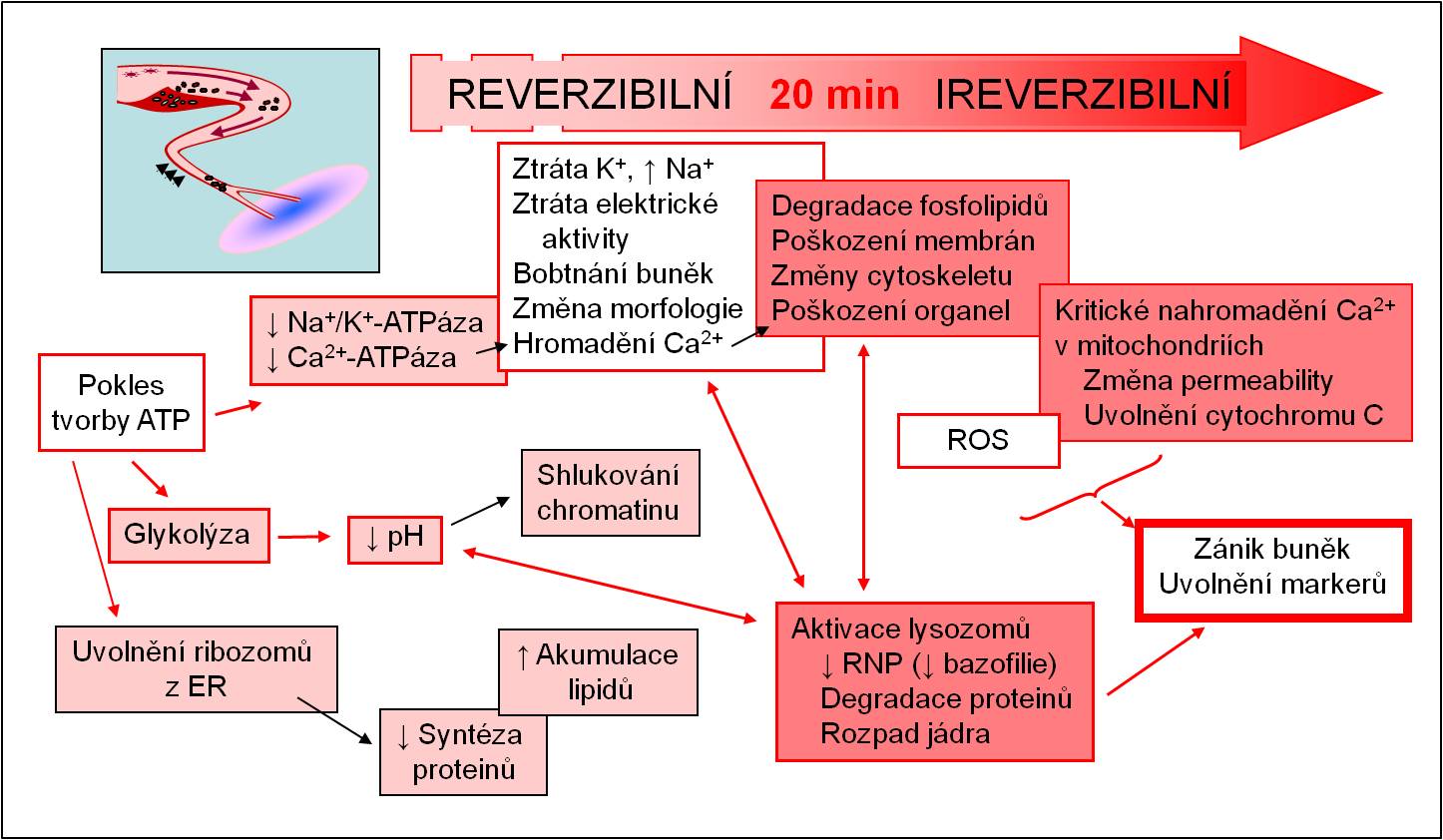
Obr. 6. Schéma nitrobuněčných změn v ischemizovaných svalových buňkách myokardu.
Doba přežití buněk myokardu při dosažení kritické ischémie po úplném uzávěru věnčité tepny je 20 minut (obr. 7). Do té doby je poškození reverzibilní – je zachována schopnost regenerace, a pokud se obnoví dodávka kyslíku, obnoví se i funkce vlákna. Tato skutečnost má prvořadý klinický význam. Ukazuje, jak důležité je co nejdříve poskytnout postižené osobě odpovídající kvalifikovanou pomoc.
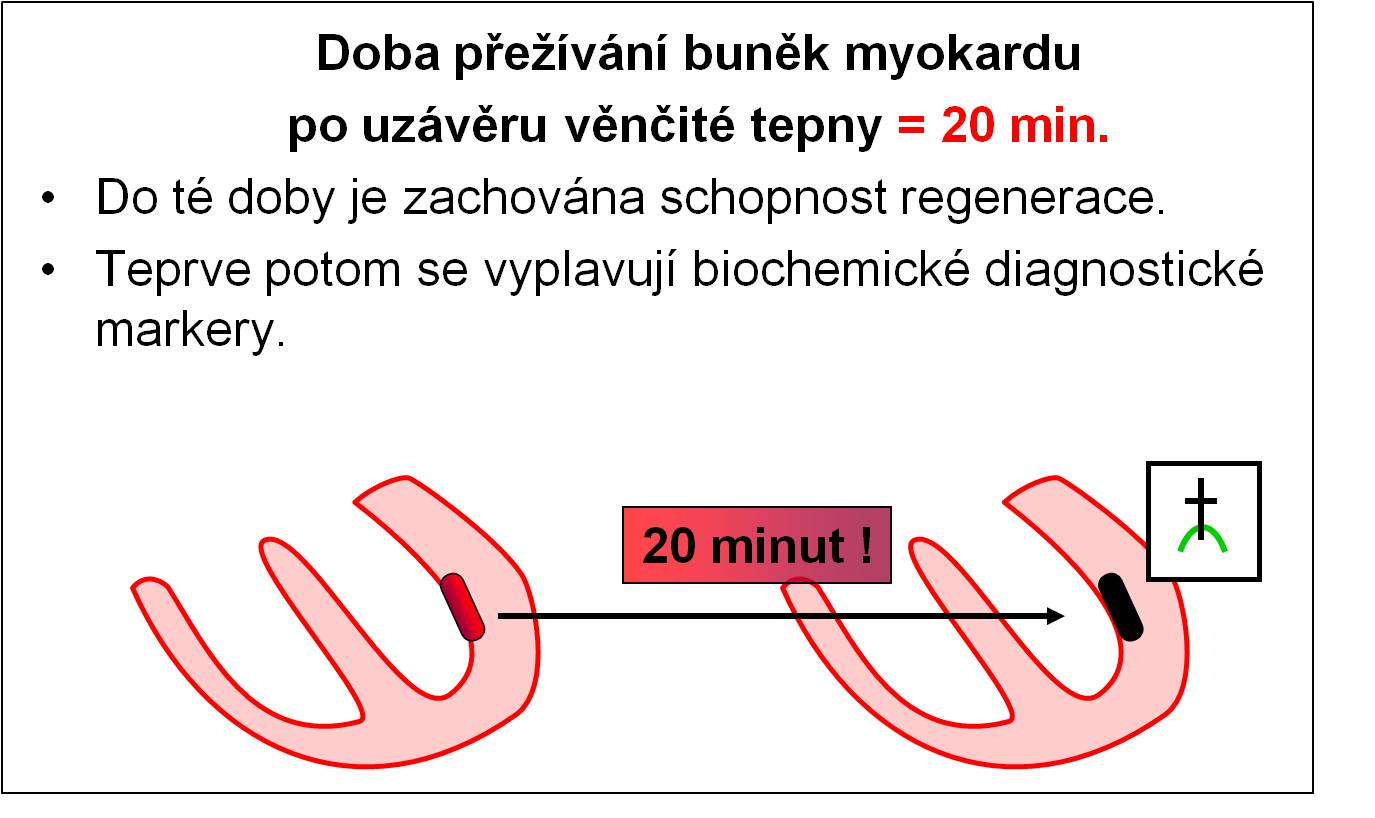
Obr. 7. Doba přežití svalových buněk myokardu po dosažení kritické ischémie.
Další klinicky důležitá skutečnost spočívá v tom, že nekróza obvykle nezasahuje celé ložisko a celou tloušťku myokardu najednou. Obvykle se šíří od endokardu k epikardu a od centra povodí příslušné tepny k periférii. Průměrně trvá 6 hodin, než nekróza prostoupí od endokardu tloušťkou celé stěny (4 – 12, výjimečně 24 hodin) (obr. 8). Velká část vláken tak může být včasnou léčbou zachována. I tato okolnost ukazuje na mimořádnou důležitost včasné odborné pomoci. Šance na přežití co největšího počtu vláken jsou tím větší, čím dříve se obnoví průchodnost koronární cévy a dodávka kyslíku. To zmenšuje rozsah následné nepříznivé remodelace postižené komory.
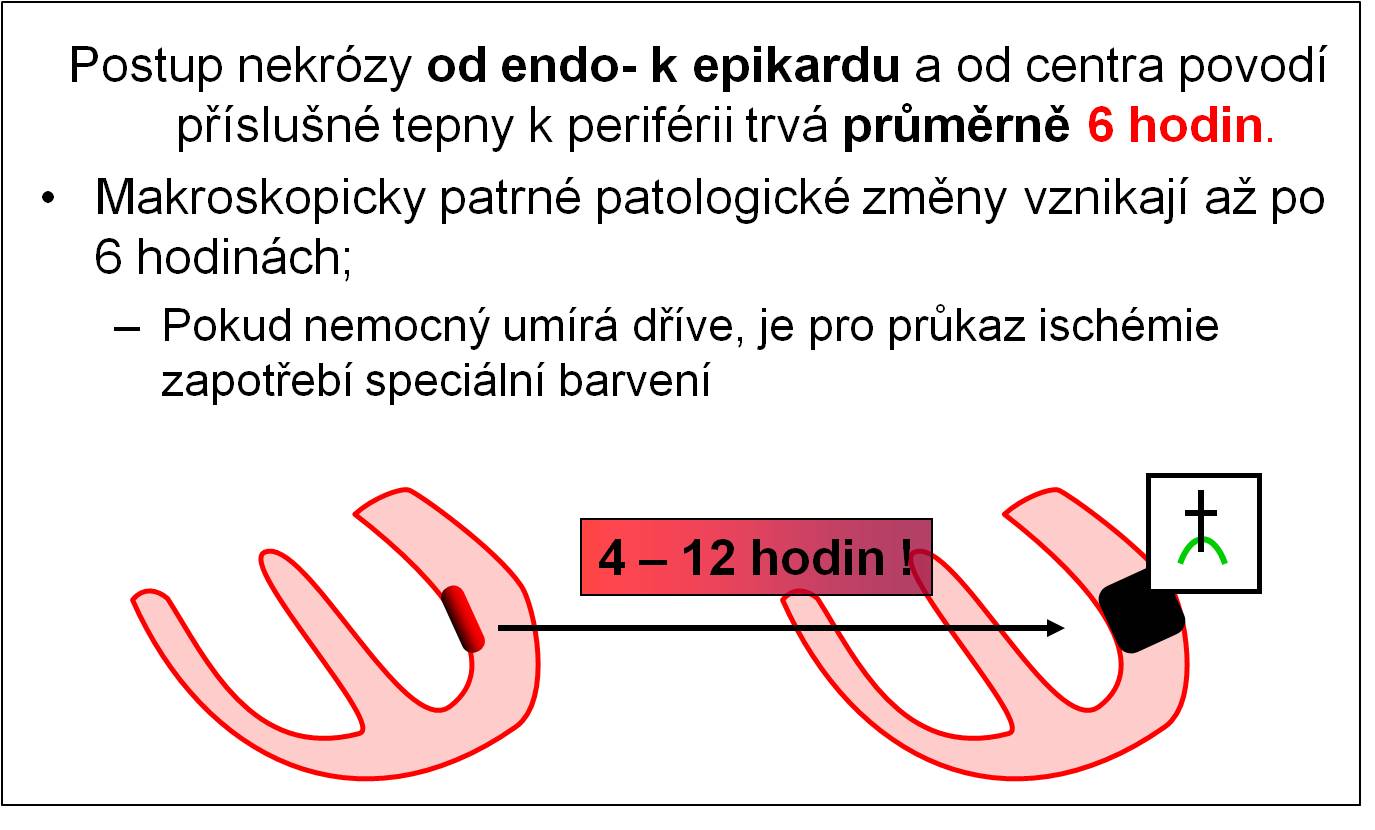
Obr. 8. Postup nekrózy stěnou myokardu – od vnitřních vrstev k zevním.
| Čas | Změny |
| 20 s | Pokles ejekční frakce, pokles arteriálního tlaku, bolest, úzkost Aktivace sympatiku: studený pot, tachykardie, bledostPočáteční změny v EKG |
| 20 min | Doba přežívání buněk: zánik buněk |
| 2 – 4 hod | Vzestup markerů v periferní krvi |
| 6 – 12 hod | Prostup nekrózy stěnou myokardu |
Tabulka 4: Shrnutí hlavních klinicko-patologických změn v ischemizovaném myokardu.
Dysfunkce myokardu
Časná dysfunkce komory se rozvíjí za 5 – 6 s po uzavření koronární cévy a projevuje se následovně (obr. 9 a 10):
- Hypokineza až akineza – snížení až vymizení kontrakcí v ischemickém ložisku;
- Dyskineza – paradoxní systolické vyklenování v postižené části myokardu;
- Hyperkineza – kompenzační zesílení kontrakcí svaloviny v okolí postižené části myokardu.

Obr. 9. Následky časné dysfunkce komory.
Hlavním následkem časné dysfunkce je pokles ejekční frakce (EF) a srdečního výdeje. K poklesu ejekční frakce dochází už za 15 s po uzavření koronární arterie. Klasifikuje se následovně:
- EF > 55 % = normální;
- EF 45-55 % = lehce snížená;
- EF 35-45 % = výrazně snížená;
- EF < 35 % = velký rozsah infarktu, špatná prognóza;
- EF 15-20 % = hodnoty typické pro kardiogenní šok.
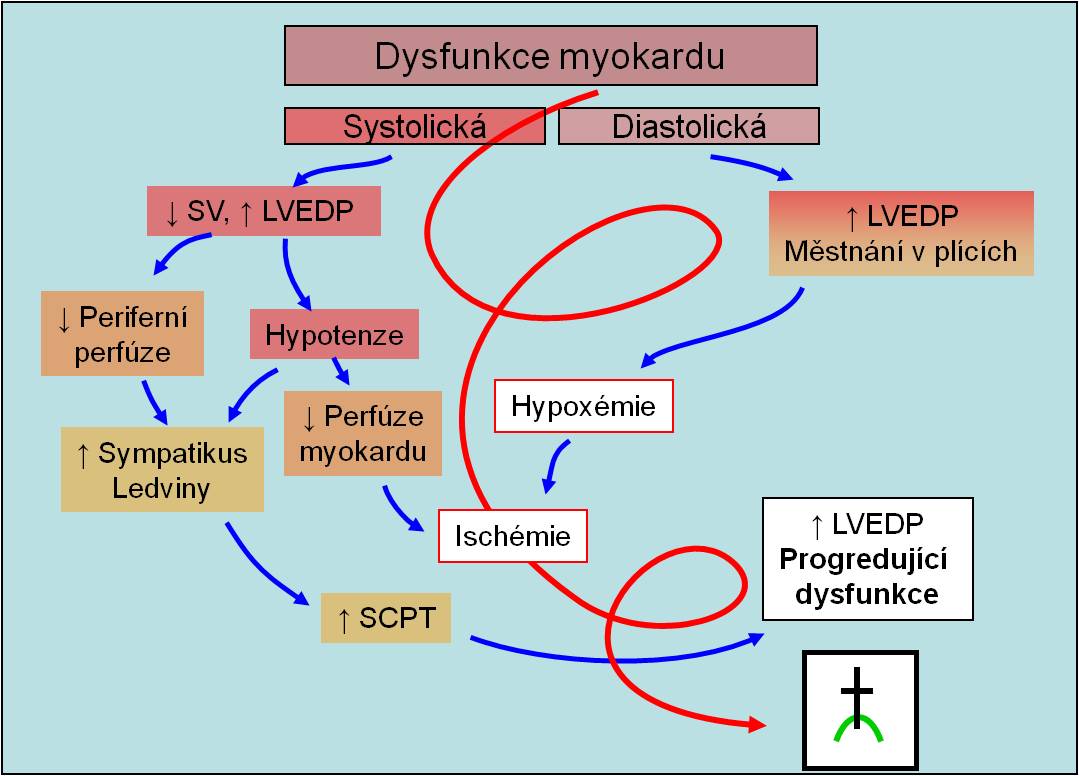
Obr. 10. Časná dysfunkce a spirála kardiálního šoku. Podle: Harrison’s Principles of Medicine, 15th Ed. McGraw-Hill, 2001.
Etiologie ischémie myokardu
Přehled příčin ischémie myokardu (obr. 11):
- Ateroskleróza:
- Vůbec nejčastější bezprostřední příčinou akutní ischémie je vznik trombu na aterosklerotickém podkladě;
- Ateroskleróza tak je příčinou zúžení a ischémie v 95 % všech případů ICHS;
- Může způsobovat až nekrózu (infarkt).
- Vzácně:
- Spasmy koronárních cév;
- Embolie;
- Arteritis;
- Velice vzácně – trombóza v koronární tepně bez aterosklerózy.
- Rovněž hypoperfúze myokardu při šoku může vést k hypoxii. Zvyšuje se objem svalových vláken (intracelulární otok). V pokročilých fázích se mohou objevovat subendokardiální nekrózy. Po funkční stránce klesá poddajnost srdeční stěny a klesá ejekční frakce.

Obr. 11. Příčiny ischémie myokardu.
Postup akutní okluze koronární arterie lze popsat následovně:
- Aktivace trombózy:
- Lokální dysbalance faktorů přítomných v lumen cévy anebo v její stěně;
- Podíl endotelové dysfunkce (snížení NO, PGI2);
- Podíl zánětu (v aterosklerotickém ložisku);
- Mikroskopická fisura, ruptura nebo exulcerace plátu;
- Aktivace destiček; agregace destiček (zvýšení TxA2, vazba fibrinu na glykoproteinový receptor GP IIb/IIIa trombocytů); primární zátka;
- Koagulace; fibrinová zátka;
- Neúplně obturující trombus;
- Lokální dysbalance faktorů přítomných v lumen cévy anebo v její stěně;
- Úplně obturující trombus;
- Pamatovat na účast spasmu postižené artérie.
Hypoperfúze a ischémie ledvin
Fyziologické základy
Ledviny dostávají téměř 1/4 (22 %) srdečního výdeje, a v tomto ohledu tedy jsou na druhém místě za játry (27 % srdečního výdeje). Po přepočtu na jednotku hmotnosti ovšem zjišťujeme, že ledviny jsou krví zdaleka nejlépe zásobeným orgánem v těle (3,5 ml/g tkáně/min; játra asi 1,0 ml/g tkáně/min).
Ledvinový parenchym nepotřebuje toto množství krve pro zajištění základní životaschopnosti buněk, ale pro výkon funkcí. Hustota mitochondrií v tubulárních buňkách dosahuje hustoty mitochondrií v neuronech. Proximální tubuly, bohatě vybavené mitochondriemi, jsou mnohem vnímavější k hypoxii než tubuly distální, jejichž pracovní zatížení je menší. Krevní tlak v glomerulárních kapilárách je nezvykle vysoký (kolem 60 mm Hg). Hlavním odporovým článkem ledvinové cirkulace jsou eferentní arterioly – odpovídají za více než 40 % jejího cévního odporu.
Lokální autoregulace zajišťuje, že krevní průtok ledvinami je v širokém rozmezí změn arteriálního tlaku konstatní – v rozmenzí mezi 75 – 160 mm Hg arteriálního tlaku jsou změny průtoku pouze několik procent. Neméně efektivně, ne-li efektivněji, je v uvedeném tlakovém rozmezí regulována i rychlost glomerulární filtrace.
Etiologie ischémie ledvin
Ke snížení průtoku a. renalis dochází velmi často. Ledviny mohou být poškozeny sníženou dodávkou krve z důvodu snížení srdečního výdeje (hypovolémie, šok) anebo z důvodu cévního onemocnění (např. ateroskleróza renální tepny). Nejčastější příčinou je redistribuce krve při centralizaci oběhu při pokročilém srdečním selhávání (kardiorenální syndrom), šokových a dalších stavech (dehydratace u starších lidí), kdy působením vyplavujících se katecholaminů a angiotenzinu dojde ke spasmu drobných intrarenálních vas afferens i efferens a ledvina (zejména její korová část) je ischemizována. Navíc se velmi často lokálně uplatňují progredující aterosklerotické (a následně trombotické) uzávěry. Akutním stavem je např. embolie přívodných renálních tepen (ateromové pláty z aorty anebo srdeční tromby).
U všech těchto stavů pak dochází ke globální (neselektivní) poruše ledvin vlivem poklesu glomerální filtrace, která se může manifestovat obrazem akutního prerenálního oligurického až anurického ledvinového selhání. Ischémie může vést k ischemické nekróze tubulů.
Patofyziologie ischémie ledvin
Ledviny reagují na hypoperfúzi produkcí reninu, vytváří se angiotenzin. Angiotenzin II zužuje jak periferní arterioly, tak vény a přímým účinkem na epitely proximálních tubulů mimořádně účinně stimuluje reabsorpci soli a vody v ledvinách. Jeho působení je kritické pro udržení objemu tekutin a náplně cirkulace. Odpověď s účastí angiotenzinu se ale rozvíjí se zpožděním za odpovědí sympatiku. Začíná asi po 10 minutách a dosahuje plného účinku asi za 1 hodinu.
Pokud postižení není kritické, rozvíjí se ischemická nefropatie (při chronicky sníženém přívodu krve se může vyvíjet i po dobu několika let). Vystupňovaná vazokonstrikce v ledvinách, provázející poplachovou redistribuční reakci, dovede omezit průtok krve parenchymem na 1/2 normálního množství. Na rozdíl od jiných tkání se ledviny dovedou přizpůsobit poklesu krevního zásobení až do 1/5 normálních hodnot. S poklesem množství přiváděné krve klesá rychlost glomerulární filtrace, a s ní i nálož iontů Na+, vody a ostatních složek, které se z tubulů transportují zpět do krve. Proto se při omezení přítoku krve a při cirkulačním selhání při šoku, MODS anebo SIRS zároveň snižuje i spotřeba kyslíku v ledvinách. Pokud je omezení přísunu krve těžké, glomerulární filtrace prakticky ustane a buňky spotřebovávají kyslík jen pro své přežití.
Při poklesu přívodu krve pod uvedenou hranici 20 % normální úrovně buňky čelí kritické hypoxii a po několika hodinách umírají. Nejcitlivější k nedostatku kyslíku jsou tubulární buňky bohaté na mitochondrie – zejména buňky proximálního tubulu, silného vzestupného raménka Henleovy kličky a proximálního sběrného kanálku na rozhraní mezi kůrou a dření. K hypovolémii a vazokonstrikci, omezujícím dodávku kyslíku, se navíc přidává intraparenchymový edém. Může se rozvinout akutní tubulární nekróza – původní prerenální poškození pak pokračuje jako intrarenální. Odloupané epitelie ucpávají kanálky (obr. 12). Výkon ledvinových funkcí potom spočívá na činnosti zbývajících méně poškozených nefronů. Je vhodné mít na paměti, že termín akutní tubulární nekróza by se měl používat pouze v případech ověřených renální biopsií; u kriticky nemocných pacientů, kterých se toto téma dotýká nejvíce, toto vyšetření často nelze provést.
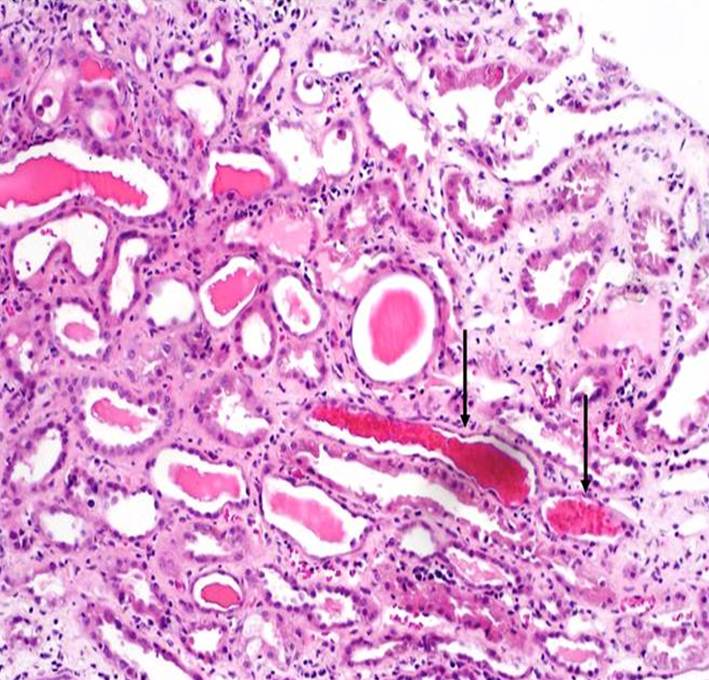
Obr. 12. Akutní tubulární nekróza – histologický obraz. Na snímku jsou v některých tubuolech zachyceny hemosiderinové válce (šipky) a jednotlivé deskvamované epitelie (horní třetina snímku). Edém intersticia je mírný s nevýraznou lymfocytární infiltrací. Barvení HE, zvětšení 100x. Dokumentaci laskavě poskytl MUDr. K. Krejčí, PhD, 3. interní klinika – nefrologická a revnatologická LF UP a FN v Olomouci.
Pokud je frakce fungujících nefronů příliš malá, dochází k manifestnímu selhání ledvin. Projevem je oligurie a s ní spojený nárůst kreatinémie a koncentrací dalších N-katabolitů, nárůst objemu tělních tekutin (objemové přetížení oběhu), metabolická acidóza, hyperkalémie. Pacient může zemřít během jednoho nebo dvou týdnů.
Nefrony ucpané epiteliemi se na tvorbě moči mohou začít podílet až dlouho po odeznění prvotní příčiny nekrózy. Odloupané epitelie jsou pomalu odklízeny a uvolňují lumen kanálků. Namísto buněk epitelu lemuje lumen kanálků obnažená bazální membrána, takže primární moč, která do kanálků vstupuje, jimi protéká bez úpravy (chybí koncentrační funkce). Bazální membrána se však postupně rekolonizuje ze zachovaných ostrůvků epitelových buněk, které proliferují, kloužou po bazální membráně a ze stran zakrývají defekty. Podmínkou pro zdárný průběh tohoto procesu je zachovaná integrita bazální membrány. Doba potřebná pro kolonizaci se udává mezi jedním a čtyřmi týdny, konečná úprava funkcí však nastává po měsících až jednom roce.
Hypoxie a ischémie gastrointestinálního traktu (GIT)
Hypoperfúze spojená s hypoxií žaludeční sliznice v těžkých stavech bývá příčinou vzniku stresových vředů, které vážným způsobem komplikují stav nemocného.
Navíc jsou hypoxie a ischémie GIT jsou provázeny poruchami střevní bariéry. Porušení bariéry vede k průniku toxinů a bakterií do portální krve a do systémového oběhu. Rozvíjí se toxémie, sepse a spirála smrti nevratného poškození organismu. Střevo se právem označuje jako „motor sepse„.
Hypoperfúze a ischémie jater a pankreatu
Fyziologické základy
Arteriální tepna obstarává 20 % zásobení jater a přináší kyslík. Portální krev obstarává 80 % zásobení jater a přivádí vstřebané živiny. Portální vénou do jater přichází asi 1500 ml krve za minutu ze žaludku, tenkého a tlustého střeva a sleziny (obr. 13) při nízkém hydrostatickém tlaku (5 – 10 mm Hg).
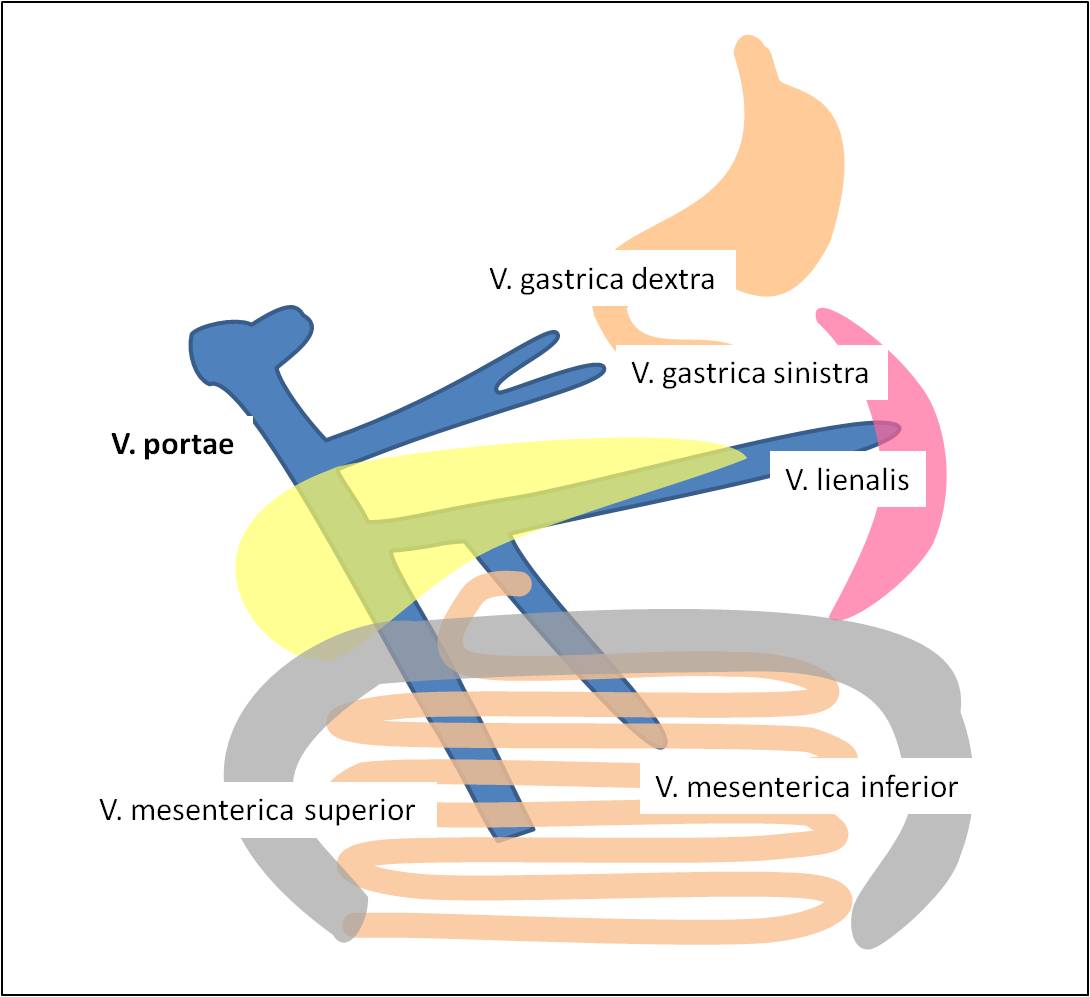
Obr. 13. Venózní zdroje v. portae.
Patofyziologie ischémie jater
Proud v portální žíle se vzhledem k nízkému tlaku zpomaluje při každém zvýšení odporu, na který krev na své cestě játry do dolní duté žíly narazí. Potom ve vena portae vzniká portální hypertenze (> 12 mm Hg). Vena portae je za primární kapilární sítí, takže portální hypertenze se přímo nedotýká tlaku v systémové cirkulaci. Přehled příčin portální hypertenze podává tabulka 5.
| Přehled hlavních příčin portální hypertenze | ||
| Zvýšený cévní odpor | Prehepatické příčiny | Trombóza portální vény nebo jejích přítoků |
| Utlačení žíly vnějším útvarem (např. nádorem) | ||
| Hepatické příčiny | Jaterní fibróza anebo cirhóza | |
| Infekce, napadení parazity | ||
| Posthepatické příčiny | Omezení průchodnosti, např. trombóza žil vedoucích z jater (Budd-Chiariho syndrom) | |
| Chronické selhání pravého srdce | ||
| Konstriktivní perikarditida | ||
| Zvýšený průtok krve portálním řečištěm | Arterio-portální zkraty | |
Tabulka 5. Přehled hlavních příčin portální hypertenze.
Porušené proudění krve játry a portální hypertenze ve velké míře spoluurčují symptomatologii postižení jater. Samy o sobě dokonce přinášejí těžké komplikace také v případě, že se objeví i bez průvodního jaterního onemocnění. Vyvolávají jaterní hypoxii, která sama může vést až k zániku buněk, zvýšení plazmatických indikátorů a – při chronickém postižení – k rozvoji fibrózy a cirhózy.
K ischemickému postížení jater ovšem může dojít i naprosto nezávisle na portální hypertenzi při hypoperfúzi a hypoxii jater při selhání oběhu. Postižení hepatocytů se morfologicky může manifestovat centrilobulárními nekrózami. Uvádí se však, že selhání jater při akutním selhání cirkulace bývá vzácné.
Ischemická ložiska se při hypoperfúzi rovněž mohou objevit v pankreatu.
Periferní tkáně a svalstvo při hypoxii a ischémii
Při hypoperfúzi se ve tkáních trpících hypoxií rozvíjí anaerobní metabolismus. Hromadí se laktát. Z nedostatku ATP selhávají iontové pumpy a zanikají iontové gradienty na membránách. Z buněk do intersticia vystupují K+ ionty. Do buněk vstupují Ca2+ ionty a spolu s ionty H+ a dalšími faktory spouštějí procesy zániku buněk – autofágii, apoptózu a nekrózu. Aktivují se děje proteolýzy, lipolýzy a degradace nukleových kyselin.
Výrazná hypoperfúze a hypoxie kosterního svalstva vede ke zhoršení jejich funkce a výkonnosti. To může být kritické v připadě postižení dýchacích svalů a bránice.
Cévní stěny při hypoxii a ischémii
Štěpením buněčných struktur ve tkáních vznikají biologicky aktivní peptidové a lipidové produkty, které se hromadí ve tkáních. Vznikají reaktivní kyslíkové radikály. To všechno negativně ovlivňuje stav hladkého svalstva cév.
Aktivace endotelu je fyziologickou složkou cévní odpovědi na hypoxii a ischémii. Dochází k posunům v syntéze a uvolňování NO a dalších mediátorů. Zároveň se mění adhezní vlastnosti endotelu. Je alterována permeabilita kapilár. Ve tkáních dochází k hemokoncentraci. Produkty rozpadu tkání, nadměrně uvolňovavé zánětové mediátory a mediátory aktivovaného endotelu mohou způsobit, že vratná aktivace endotelu přejde v generalizovanou endotelovou dysfunkci, která už obvykle provází rozvoj SIRS. Následkem může být tvorba mikrotrombů a vznik DIC. Nejtěžší komplikací DIC je tzv. purpura fulminans, při níž vznikají rozsáhlé kožní a slizniční nekrózy a následně i gangrény; často se pozoruje právě při sepsi.
Zpracoval: Jaroslav Veselý, Ústav patologické fyziologie LF UP v Olomouci. Kazuistiku Difúzní edém po celkové hypoxii zpracoval prof. MUDr. Miroslav Heřman, PhD, Radiologická klinika LF UP a FN v Olomouci.


