

Definice a terminologie
Hypertenze, přesněji systémová arteriální hypertenze, je charakterizována chronickým zvýšením systémového arteriálního krevního tlaku nad normální hodnoty.
| Měření tlaku | Systolický tlak (mm Hg) | Diastolický tlak (mm Hg) |
| Ve zdravotnickém zařízení | ≥ 140 | ≥ 90 |
| 24hodinové monitorování | ≥ 125 | ≥ 80 |
| V domácích podmínkách | ≥ 135 | ≥ 85 |
Tabulka 1. Hraniční hodnoty systémového arteriálního krevního tlaku (mm Hg) podle podmínek měření.
| Klasifikace | Systolický tlak (mm Hg) | Diastolický tlak (mm Hg) |
| Optimální | < 120 | < 80 |
| Normální | 120 – 129 | 80 – 84 |
| Vysoký normální | 130 – 139 | 85 – 89 |
| Hypertenze 1. stupně („mírná“) | 140 – 159 | 90 – 99 |
| Hypertenze 2. stupně („středně závažná“) | 160 – 179 | 100 – 109 |
| Hypertenze 3. stupně („závažná“) | ≥ 180 | ≥ 110 |
| Izolovaná systolická hypertenze | ≥ 140 | < 90 |
Tabulka 2. Kategorie hladin systémového arteriálního krevního tlaku.
Podle znalosti, nebo neznalosti příčiny hypertenze se systémová arteriální hypertenze klinicky dělí na tzv. primární neboli esenciální hypertenzi a sekundární hypertenzi.
Tzv. primární (esenciální) hypertenze zahrnuje asi 95 % všech případů. Nejde však o jediné onemocnění, dokonce ani o jednotnou skupinu. Řadí se sem různé syndromy lišící se etiologií a patogenezí. Dosud tradovaná fráze, že původ primární hypertenze je naprosto neznámý, dnes už neodpovídá skutečnosti. Za všechny formy hypertenzní nemoci jsou odpovědné poruchy ledvin. S výjimkou tzv. izolované pružníkové hypertenze není známa žádná jiná forma systémové arteriální hypertenze, která by nevznikla následkem poruchy ledvinových funkcí.
Plicní hypertenze a portální hypertenze mají ve srovnání se systémovou arteriální hypertenzí zcela odlišnou etiologii i patogenezi. Tato odlišnost především vyplývá z nepřítomnosti tlakového přepadu v plicním anebo portálním řečišti.
Epidemiologie hypertenze
V industrializovaných zemích západního typu došlo zejména v posledních asi 50 – 70 letech k prudkému nárůstu prevalence hypertenze („bílá smrt“ 20. století). Platí to i pro další civilizační choroby, jako jsou ostatní kardiovaskulární a cerrebrovaskulární onemocnění, nádory, obezita, Alzheimerova nemoc, diabetes mellitus, osteoporóza a mnohé další.
Prevalence hypertenze v ČR
Prevalence chronické systémové tepenné hypertenze u dospělých obyvatel v naší republice je dle nejnovějších dat 43,6 %; u mužů 50,2 %, u žen 37,2 % (Cífková a spol., J. Hypertens. 28, 196-203, 2010).
Rizikové faktory hypertenze
Rizikové faktory hypertenze lze rozlišit na dvě skupiny:
- První skupina zahrnuje exogenní faktory, tzn. nedědičné.
- Druhá skupina zahrnuje faktory, které jsou dány primárně a lze je jen stěží ovlivnit. Patří k nim především dědičné predispozice, věk a pohlaví.
Hlavními exogenními rizikovými faktory jsou faktory našeho životního stylu, k nimž hlavně počítáme:
- Faktory úzce související s nevyhovující stravou:
- Obezita;
- Vysoká spotřeba kuchyňské soli (NaCl);
- Nedostatečnou fyzickou aktivitu.
Dalšími exogenními rizikovými faktory jsou:
- Psychosociální stres;
- Spotřeba alkoholu;
- Kouření.
Z výčtu faktorů vyplývá, že hlavními rizikovými faktory hypertenze jsou nevyhovující strava a s ní související nevyrovnaná energetická bilance. Odhaduje se, že strava se na zdravotním stavu obyvatelstva žijícího v industrializovaných zemích podílí 30 – 80 %. Pro populaci v ČR se tento podíl oceňuje asi na 60 %. Znamená to, že ze všech vnějších faktorů má strava největší dopad na zdraví lidí.
Obezita
Obezita je hlavním rizikovým faktorem hypertenze. O patogenetickém vztahu mezi obezitou a vznikem hypertenze vypovídá skutečnost, že 15 – 35 % obézních má hypertenzi. U neobézních osob se hypertenze vyskytuje asi v 8 – 10 %. Obezita se činí odpovědnou až za 78 % rizika esenciální hypertenze u můžů a 65 % rizika u žen. Nadváhu má téměř 75 – 80 % osob s primární hypertenzí. Hypertenze daleko častěji provází přírůstek hmotnosti s věkem než obezitu, která trvá od mládí; z hlediska ostatních kardiovaskulárních rizik ale má obezita v mladším věku horší prognózu než obezita vzniklá později. O etiopatogenetickém působení obezity na hypertenzi můžeme usuzovat hlavně proto, že poškození mikrocirkulace arterioskleróhzou a parenchymu metabolickými faktory může sekundárně alterovat ledvinovou funkční křivku. Obezitu navíc provází zvýšená aktivita sympatiku.
Obezita se spojuje s četnými dalšími metabolickými poruchami, které všechny vystupují jako rizikové faktory kardiovaskulárních onemocnění. Soubor byl pojmenován syndrom X či metabolický syndrom :
- Dyslipidémie;
- Porucha glukózové tolerance;
- Hyperinzulinémie;
- Hyperglykémie;
- Diabetes mellitus;
- Hypertenze;
- Hyperurikémie;
- Trombofilie;
- Nealkoholická jaterní steatóza;
- Hirsutismus a některé další.
Spotřeba soli
Názor, že vysoká spotřeba soli je druhým nejvýznamnějším etiologickým činitelem ve významné části případů esenciální hypertenze, se opírá o následující argumenty:
- Hypertenze je běžná ve všech populacích s vysokou spotřebou soli a její prevalence v nich se zvyšuje s věkem. V těchto populacích mají jedinci, kteří v průměru konzumují stejné dávky soli, hodnoty krevního tlaku velice rozptýleny.
- Naproti tomu hypertenze je prakticky neznámá v populacích, kde se do potravy nepřidává sůl a spotřeba NaCl je menší než 50 mmol/den (tj. < 3 g/den). Tyto populace jsou navíc obvykle vysoce fyzicky aktivní a přijímají – někdy nedostatečně – potravu, která může objemná, ale je energeticky málo bohatá a má vysoký obsah tzv. protisodíkových iontů, zejména K+ a Mg2+. Průměrný arteriální krevní tlak v takových populacích je 100/65 mm Hg a s věkem se téměř nezvyšuje. Přitom meze variací tlaku jsou daleko užší, než je běžné v populacích se střední (125 – 300 mmol/den) a vysokou (nad 300 mmol/den) spotřebou soli.
- V komunitách, kde se hypertenze původně nevyskytovala, se signifikantně zvyšuje medián arteriálního krevního tlaku poté, co se přestěhovaly do prostředí, kde se strava, kulturní zvyky a způsob života blíží západním. U přistěhovalců, kteří takto pozbyli ochranné působení původního prostředí, se potom vývoj hodnot arteriálního tlaku rychle přizpůsobuje statistikám starousedlíků. Analýzy ukazují na podstatný podíl dávek soli. Tento závěr významně podporují i výsledky studia vývoje hypertenze u šimpanzů.
- V populacích konzumujících mimořádná množství soli dosahuje prevalence hypertenze až 40 %. Tak je tomu např. u obyvatel severních japonských ostrovů, kde je průměrný denní příjem soli kolem 435 mmol (asi 25 g/den).
- Člověk intermitentně přijímá sůl a vodu a starost o homeostázu přenechává svým ledvinám. Právě ledviny jsou orgánem odpovědným za rezistenci či citlivost k soli a vývoj hypertenze. U jedinců senzitivních k soli je k vyloučení nadměrných dávek Na+ ledvinami nutný vyšší perfúzní, a tedy arteriální tlak.
Sůl konzumujeme v průměrných denních dávkách kolem 10 – 15 g. Až 80 – 85 % tohoto množství dostáváme v hotových potravinách. Současné patofyziologické a klinické poznatky ukazují, že ledviny přibližně jedné čtvrtiny osob naší populace se s tak velkou chronickou zátěží sodnými ionty nedovedou vypořádat.
Příjem soli v evolučních souvislostech
Lidé jako rod jsou na Zemi tři a půl miliónu let. Z toho 99,8 % času přijímali potravu s nízkým obsahem Na+ iontů, vysokým obsahem iontů K+ a malou energetickou hutností. Jestliže uznáváme Darwinovy principy za platné, můžeme z nich logicky odvodit, že lidský organismus je adaptován na dietu s nízkým obsahem soli. E. Ginter a jiní uvádějí, že naši předkové žijící v pozdním paleolitu před 30 000 – 50 000 lety přijímali jen 30 mmol Na+ (1,8 g soli) denně, ale až 285 mmol K+ denně. Dnešní spotřeby jsou asi 170 - 260 mmol Na+ (asi 10 – 15 g soli) a pouze 64 mmol K+ za den. To znamená, že naše dnešní dávky Na+ jsou asi 5 – 10x vyšší a dávky K+ zároveň asi 4x nižší než u našich nedávných předků. Dřívější strava byla navíc objemná, měla vysokou výživnou hodnotu, ale měla malou energetickou hustotu. Její trávení bylo pomalé a provázené jen mírnou postprandiální sekrecí inzulínu.
Někdy se evoluční hlediska zpochybňují poukazem na to, že sice mohou odůvodnit citlivost k Na+, ale nedovedou vysvětlit, jak se v prostředí chudém na NaCl mohla u našich předků vyvinout schopnost tolerovat velké dávky soli (tzn. rezistence k soli). Tuto schopnost nesporně vykazuje velká část současné lidské populace, jak tomu nasvědčují nyní konzumovéné dávky soli. Podobná argumentace však nejspíše vyplývá z nedorozumění. Evoluční selekční tlaky neeliminují neutrální, ale nepříznivé vlohy. Pokud vznikaly mutace schopné uchovat NaCl v těle, nemohly se v prostředí chudém na sůl nijak nepříznivě uplatnit. Při nízkém příjmu NaCl se chovaly neutrálně, a proto chyběl tlak na jejich eliminaci. Naopak, jedinci schopní šetřit sůl, byli zvýhodněni. Dnes se při vysoké spotřebě soli tato výhoda ukazuje jako fatální. Znovu máme co dělat se „spořivými vlohami“ analogickými těm, které vynesl na výsluní výzkum obezity a diabetu typu 2.
Poznatek, že složení přijímané potravy, a v ní obsah sodných iontů, je podstatným kauzálním faktorem hypertenze, se prosazuje jen velmi zvolna. Klinická a patologická fyziologie už po pět desetiletí vysvětluje patogenezi hypertenze jako poruchu bilance sodných iontů a tekutin. Tato teorie dnes nemá srovnatelnou alternativu. Její další přehlížení není namístě. Sotva někdo věří, že se plicní nádor vytratí, jakmile postižený skoncuje s kouřením. Málokdo také očekává, že aterosklerotický plát zanikne, když pacient omezí příjem cholesterolu. Přesto pro řadu kliniků zůstává fakt, že rozvinutou hypertenzi nevyléčí snížením příjmu NaCl, jedním z nejpádnějších argumentů domněle svědčících proti primární kauzální úloze vysoké spotřeby soli v rozvoji častých forem hypertenze.
J. Šobra a jiní vyslovili varování, že člověk je jediným živočišným druhem, který může svůj životní styl modifikovat téměř bez omezení – až ke svému sebezničení. Spořivé vlohy se v prostředí nadbytku přestaly chovat neutrálně. Staly se nevýhodným dědictvím a nevítaným reliktem, proti nimž stojí dva pravděpodobně nejmocnější nepříznivé faktory západního životního stylu – neadekvátní množství a nevyhovující kvalita potravy a absence fyzické aktivity. A tak člověk v době, kdy aspiruje na pozici tvora, který bude sám projektovat svůj genetický profil, má sklon zapomínat, že existuje i jiná, dlouhou evolucí prověřená cesta, jak se vyhnout konfliktu mezi genetickou výbavou a prostředím. Jde o respektování a uvědomělé vytváření takových podmínek, v nichž by škodlivé vlohy nemohly rozvinout svou účinnost. Nepříznivé vlohy, které člověk s takovou naléhavostí odhaluje, by měly být znovu vykázány do role vloh neutrálních.
Tělesná aktivita
Fyzická aktivita je dalším z klíčových faktorů životního stylu, podobně jako strava. Zvýšená fyzická aktivita průkazně zmenšuje riziko vzniku hypertenze i za přítomnosti výše uvedených rizikových faktorů. Vede ke snížení tlaku bez ohledu na to, zda je anebo není provázena snížením váhy.
Psychosociální stres
Rozvinutá stresová reakce zahrnuje tři základní stereotypy:
- Poplachovou aktivaci sympatiku včetně dřeně nadledvin s vyplavením katecholaminů a s následným rozvinutím řetězce hemokardiorespiračních, metabolických a behaviorálních (útěk-boj) odpovědí.
- Navazující rozsáhlou aktivaci neuroendokrinního systému, zejména osy hypotalamus-adenohypofýza-kůra nadledvin, která je pomalejší, ale vede k déletrvajícím změnám.
- Významnou složkou stresových reakcí je mobilizace imunitního systému.
Fyziologická stresová reakce se za dlouhá období fylogeneze vyvinula jako dobře koordinovaná somatomotorická, hormonální a metabolická kaskáda. Neovládáme ji vůlí. Je určena k rychlému zvládnutí stresové situace. Po miliony let zajišťovala přežití v extrémních fyzických podmínkách. Jejím rozhodujícím přirozeným usměrněním je fyzická aktivita. V současném stylu života se z ní převážně uplatňují jen kusé fragmenty, zlomky jejích visceromotorických a metabolických poplachových článků. V moderní společnosti, kde převažuje spíše sociopsychologická, mentální a emoční zátěž převážně spojená ne s pohybovou, ale s tonickou izometrickou svalovou aktivitou, se stresová reakce manifestuje jako částečně atavistický projev nedostatečné adaptace. Náš sedavý způsob života jen umocňuje její potenciální škodlivý efekt.
Alkohol a kouření
Nízká spotřeba alkoholu (do 30 g denně) se neprojevuje dlouhodobými změnami tlaku. Naopak, umírněná konzumace je spojována s „francouzským paradoxem“. Osoby, které konzumují více než 60 – 80 g alkoholu denně, však mají hypertenzi častěji. U 60 – 80 % chronických alkoholiků je přítomný vysoký krevní tlak. Mechanismus působení velkých dávek alkoholu na tlak není znám.
Kuřáci mívají nižší tlak než nekuřáci. Kouření však je příčinou 9 z 10 případů chronické obstrukční plicní nemoci, která je čtvrtou nejčastější příčinou všech úmrtí v zemích západního typu. Je odpovědné za 1/3 všech úmrtí na rakovinu, která je třetí nejčastější příčinou všech úmrtí v zemích západního typu. Kouření zvyšuje riziko kardiovaskulárních a cerebrovaskulárních onemocnění, trombotizací a embolizací. Jednou z kritických kombinací, výrazně ohrožujících zdraví a život, je kouření provázené užíváním perorálních antikoncepčních prostředků, zejména u žen nad 35 let.
Genetické faktory
Podíl genetických faktorů na vývoji hypertenze nelze vysvětlit modely bimodálního rozdělení, které by na jedné straně předpokládaly vyhraněně ohrožené jedince nebo skupiny a na druhé straně jedince rezistentní. Výstižné jsou naopak modely, které počítají se široce proměnnou náchylností (susceptibilitou) obyvatelstva k hypertenzi. Opírají se o předpoklad, že dědičnost hypertenze je polygenní a kumulativní, přičemž expresivita genů, které za ni odpovídají, není veliká. Předpokládá se, že různé formy hypertenze budou vysvětleny nalezením více než jednoho vzorku rizikového genového spektra. Přitom se nevylučuje, že v polygenní síti může několik genů dominovat. V interakcích genů se se vší pravděpodobností účastní epigenetické faktory.
Vysvětlení hypertenze se formuluje v rámci tzv. mozaikových teorií. Podle odhadů odpovídá za regulaci krevního tlaku asi 25 – 70 genů. Pokud se některé varianty určitých genů podílejí na celkovém hypertenzivním fenotypu příspěvkem kolem 1 mm Hg, nebo dokonce menším, neudivuje, že výsledky asociačních studií se mnohdy nepodaří zopakovat. U relativně jednoduchého genu adrenergního β2-receptoru zatím bylo popsáno na 15 variant. Tento údaj ovšem ještě nezahrnuje proteinové izoformy vzniklé posttranskripčně mechanismem alternativního sestřihu. Představa, že bude zapotřebí současně interpretovat databázi variací 25 – 70 takových genů, dává dosti přesný obraz o obtížnosti podobného úkolu.
Genetické predispozice k hypertenzi se manifestují v těsné závislosti na interakcích s množstvím vnějších faktorů. Mezi polymorfismem určitého lokusu a jeho fenotypovým projevem je řada mezičlánků, z nichž každý může být jak pod genetickou kontrolou, tak pod kontrolou okolí. Mezičlánky znesnadňují zjištění vazby mezi určitým genotypem a fenotypem úrovně arteriálního tlaku. Rozhodující zevní činitelé hypertenze zřejmě ovlivňují ani tak kvalitu uplatněné vlohy (tj. zda se nepříznivá vloha exprimuje, nebo ne), jako spíše kvantitu (tj. nakolik se exprimuje); jejich účinky se sčítají nebo násobí.
Patofyziologie hypertenze
Tlaková diuréza a úloha ledvin v patogenezi hypertenze
Neplatí fráze dosud tradovaná v učebnicích, že původ primární hypertenze je naprosto neznámý. Za všechny formy hypertenzní nemoci jsou odpovědné poruchy ledvin. S výjimkou tzv. izolované pružníkové hypertenze není známa žádná jiná forma systémové arteriální hypertenze, která by nevznikla následkem poruchy ledvinových funkcí. Názor, že původ systémové arteriální hypertenze spočívá v dosud nejasné, povšechně změněné reaktivitě systémových cév anebo srdce, je mylný. Chronickou systémovou artreriální hypertenzi nelze vyvolat ani izolovaným nárůstem periferního odporu, ani izolovaným nárůstem srdečního výdeje, ani izolovaným zmenšením kapacity cirkulace.
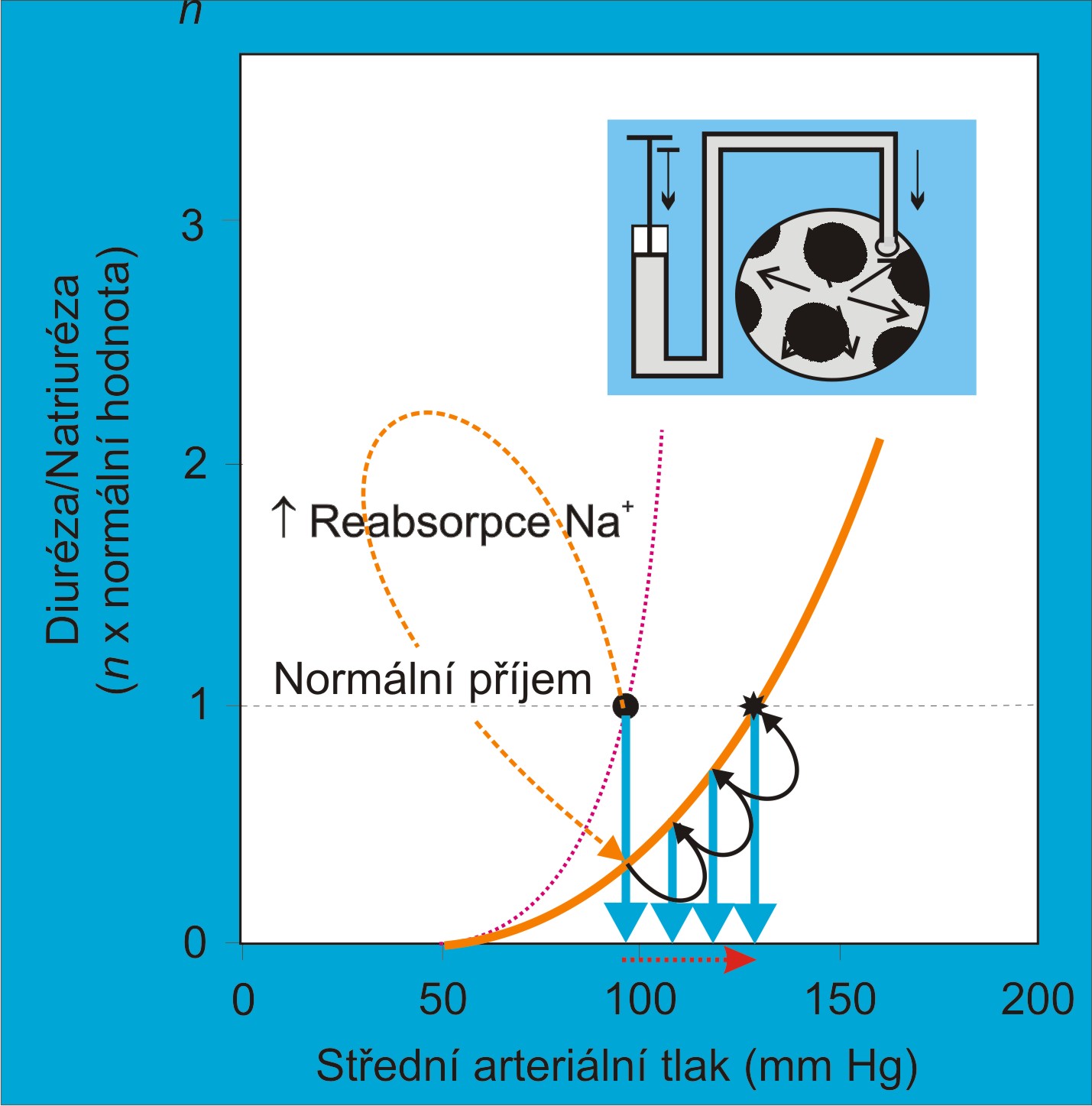
Základním funkčním ledvinovým principem je tlaková diuréza. Množství moči tvořené v ledvinách se fyziologicky zvyšuje s růstem systémového arteriálního tlaku, když se zvýší perfúzní tlak v ledvinách. Pokles systémového arteriálního tlaku naopak vede k redukci množství moči a k retenci sodných iontů a tekutin.

Obr. 1. Křivka tlakové diurézynatriurézy neboli funkční křivka ledvin. Čárkovaně je vyznačena křivka tlakové diurézy provázející zvýšenou resorpci (sníženou exkreci) sodných iontů a vody v tubulech např. při hypovolémii. (Podle AC Guyton a JE Hall: Textbook of Medical Physiology, 11th Ed. Elsevier/Saunders, Philadeplhia. 2006.) Schéma je možno zhlédnout i v animované podobě.
Hypertenze se tudíž rozvíjí jako sekundární reakce oběhu na primární poruchu ledvinových funkcí, jež zabránila zajistit vyrovnanou bilanci solí a tekutin organismu při normálním krevním tlaku. Hypertenze je symptomem takové poruchy. Proto není namístě hovořit o esenciální hypertenzi jako o nemoci s nepoznanou příčinou. Hypertenze vzniká následkem poškození ledvin. V první řadě je známkou poruchy regulace rovnováhy objemu tekutin.
Z klinických zkušeností je dobře známo, že neschopnost udržet potřebný renální perfúzní tlak v ledvinách při normálním arteriálním tlaku v cirkulaci anebo neschopnost opětovně ho zvýšit na potřebnou hodnotu vede k pozitivní iontové a vodní bilanci. Příkladem je stenóza ledvinové artérie, krajním případem je oligurické/anurické selhání anebo odnětí ledvin. Pozitivní bilance se projeví rychlým městnáním iontů a tekutin, které během několika dnů mohou přivodit srdeční selhání. Proti následkům déletrvající pozitivní bilance tekutin není organismus vybaven. Tlaková diuréza poskytuje řešení. Částečné omezení natriuretických schopností ledvin může být v určitých mezích překonáno vyšším perfúzním tlakem. Cenou, kterou je za to třeba zaplatit, je nárůst systémového arteriálního tlaku. To je podstatou systémové arteriální hypertenze.
Překonané hypotézy vzniku chronické systémové arteriální hypertenze
1. Nemožnost vyvolání hypertenze izolovaným nárůstem periferní rezistence nebo izolovaným zmešením kapacity cirkulace
Podle jedné z překonaných hypotéz vzniku hypertenze by měl být prvotní příčinou chronického zvýšení systémového arteriálního tlaku abnormální nárůst periferní rezistence a případně zmenšení kapacity krevního řečiště.
Periferní vasokonstrikce anebo centralizace oběhu jistě akutně zvýší arteriální tlak nad normu. Jsou na tom založeny krátkodobé, pohotovostní adaptační mechanismy regulace krevního tlaku. Je ale třeba mít na paměti, že déletrvající zvýšení arteriálního tlaku – pokud zůstanou nedotčeny ledvinové funkce – způsobí tlakovou diurézu.
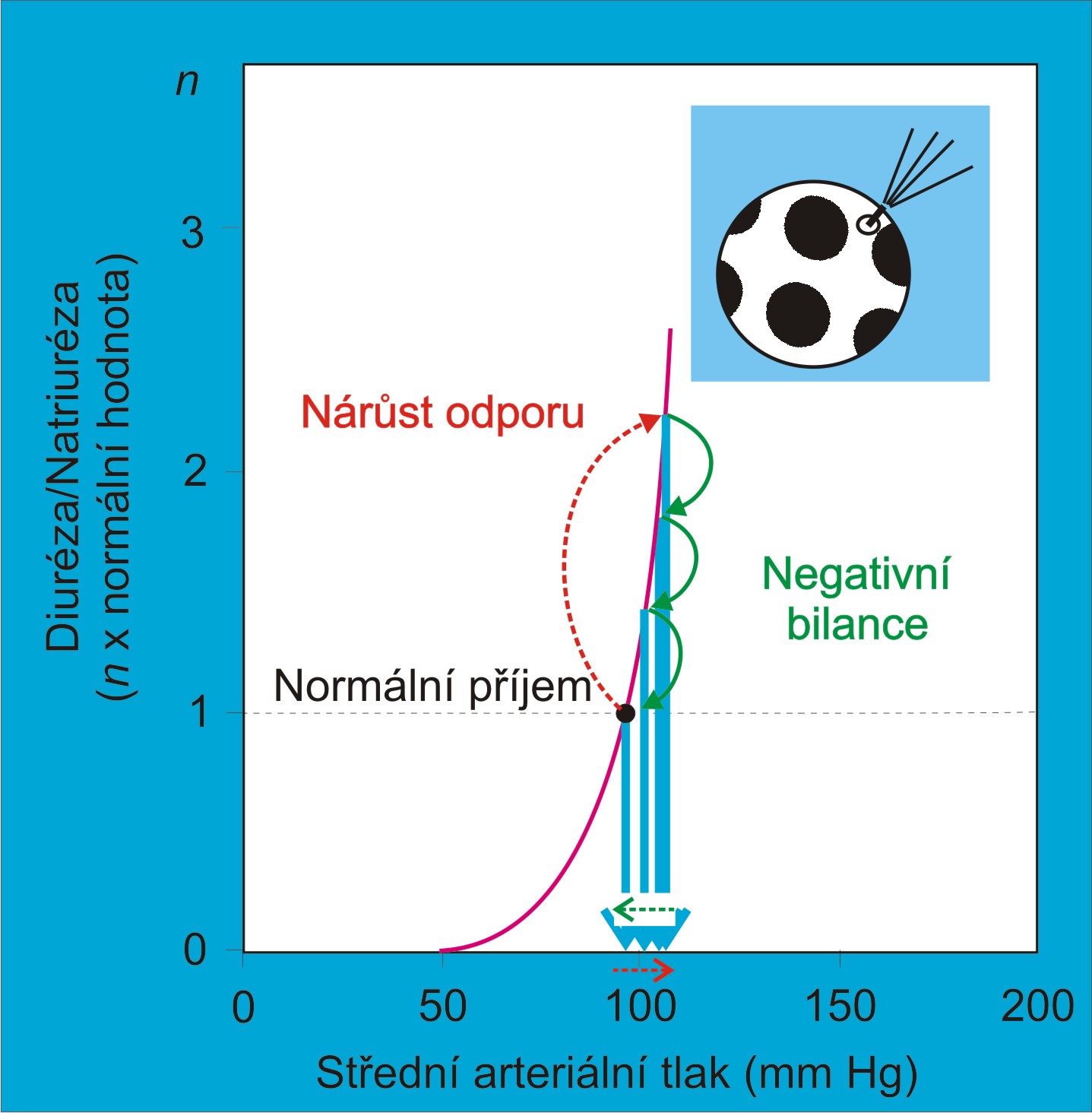
Obr. 2. Nárůst diurézy po akutním izolovaném vzestupu periferní rezistence, která způsobila vzestup arteriálního tlaku, ale neovlivnila strmost funkční křivky ledvin. Zvýšená diuréza a s ní spojená negativní bilance tekutin vrací arteriální tlak k normě. (Podle Hall JE: Am. J. Physiol. 277 (Adv. Physiol. Educ. 22), S174-S186, 1999.)
V případě generalizované vazokonstrikce mimo ledviny se zvýší arteriální tlak nad normu, ale ne dlouhodobě. Následně se rozvine tlaková diuréza a arteriální tlak se vrátí k normě. Je za to odpovědný následující sled reakcí:
- Vzroste perfúzní tlak v ledvinách;
- Vzroste diuréza;
- Ubude náplně v krevním řečišti a zmenší se střední cirkulační plnicí tlak;
- Zmenší se žilní návrat a srdeční výdej;
- Arteriální tlak poklesne na výchozí úroveň.
Jinak řečeno, diuréza uvede do souladu náplň krevního řečiště s jeho kapacitou. Zvýšený krevní tlak se po vazokonstrikci (arteriolokonstrikci anebo venokonstrikci) vrátí zpátky k normě podobně, jako když se z přehuštěného míče nebo pneumatiky odpustí vzduch. (Při poklesu tlaku pod normu nastávají opačné pochody.) Izolovaná změna celkového periferního odporu anebo kapacity cirkulace bez postižení ledvinových funkcí, které by alterovaly ledvinovou funkční křivku, tedy nemůže být příčinou chronické hypertenze. Ani dlouhodobé změny změny periferního odporu, ani dlouhodobé změny kapacity cirkulace nejsou spontánně následovány chronickými změnami tlaku, pokud ledviny pracují podle normální funkční křivky.
Uvedený závěr je podepřen celou řadou jednoznačných klinických pozorování:
- Pokud by byl systémový arteriální tlak dlouhodobě určen periferním odporem, pak bychom po chirurgickém uzavření arteriovenózního zkratu, odnětí orgánu, amputaci končetiny apod., kdy výrazně roste periferní rezistence anebo se zmenší kapacita cirkulace, měli očekávat zhoubný nárůst tlaku. K takovému nárůstu však nedochází.
- Na druhé straně mohou být hodnoty periferní rezistence u arteriovenózních zkratů, beri-beri anebo u thyreotoxikózy atp. podstatně menší než u zdravých osob. Pokud by byl systémový arteriální tlak dlouhodobě určen periferním odporem, očekávali bychom, že uvedené stavy budou provázeny výraznou hypotenzí. Hypotenze ale u jmenovaných poruch nebývá. To všechno podporuje výklad o prvořadém postavení ledvinového tlakově-diuretického mechanismu.
2. Nemožnost vyvolání hypertenze izolovaným zvýšením systolického objemu
Jiná překonaná hypotéza vysvětlovala chronické zvýšení systémového arteriálního tlaku nadměrnou stimulací myokardu anebo činnosti srdce. Absolutní podmínkou déletrvajícího zvýšení srdečního výdeje však je zvýšený žilní návrat. Toho je možno dosáhnout pouze zmenšením periferního odporu, nebo zvětšením středního systémového plnicího tlaku. Zmenšení periferního odporu by ovšem vedlo k poklesu systémového arteriálního tlaku. Zbývající možnost, chronická změna plnicího tlaku, je nemožná bez účasti ledvin. Také hypertenze domněle způsobená vysokým srdečním výdejem je tudíž zcela závislá na stavu ledvin.
Patofyziologická klasifikace chronické systémové arteriální hypertenze
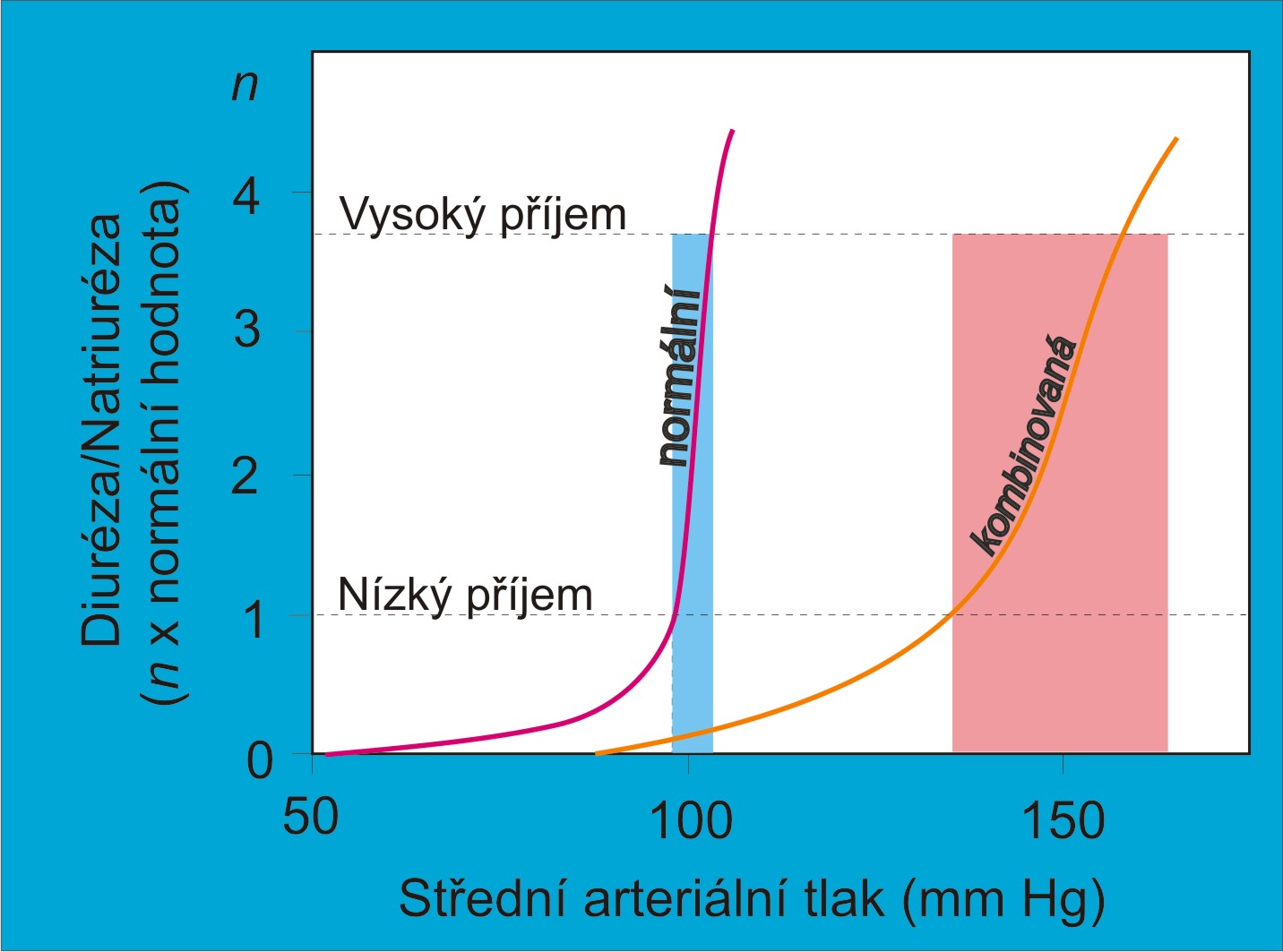
Výše uvedené příklady ukazují na postavení dlouhodobé hladiny systémového arteriálního krevního tlaku jako primární, nezávislé stavové veličiny hemodynamiky. Dlouhodobá hladina systémového arteriálního krevního tlaku není nastavena samotnou cirkulací. Je cirkulaci vnucena potřebami ledvin. Ledviny vyžadují pro plnění své základní funkce, kterou je udržování vyrovnané objemové bilance, perfúzní tlak odpovídající střednímu systémovému arteriálnímu tlaku asi 100 mm Hg. Pod touto hranicí spouštějí procesy šetření tekutinami (snížená natriuréza a diuréza, pozitivní bilance vody a tekutin), nad touto hranicí vyprazdňují tekutiny z oběhu (zvýšená natriuréza a diuréza, negativní bilance solí a tekutin), dokud se perfúzní a systémový arteriální tlak nevrátí zpátky na potřebnou hladinu.
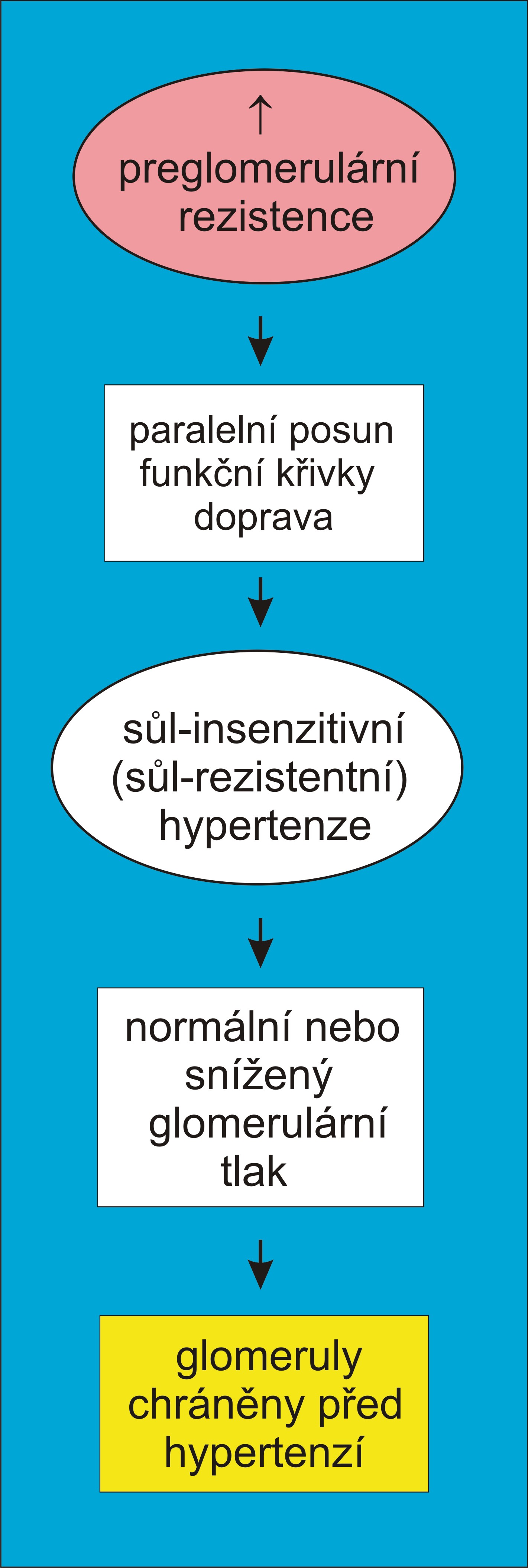
Patofyziologická klasifikace chronické systémové arteriální hypertenze se opírá o opakovaně potvrzený fakt, že není známa žádná hypertenze s normální ledvinovou funkční křivkou. Vzniku jakékoliv chronické systémové arteriální hypertenze předchází alterace křivky tlakové diuríézy. Podle tohoto kritéria se různé formy hypertenze dělí do tří skupin:
- Hypertenze objemového typu se skloněnou ledvinovou funkční křivkou, citlivou k příjmu soli;
- Hypertenze vazokonstrikčního typu s posunutou ale strmou funkční křivkou, necitlivou k příjmu soli;
- Hypertenze smíšeného typu, s posunutou a skloněnou funkční křivkou.
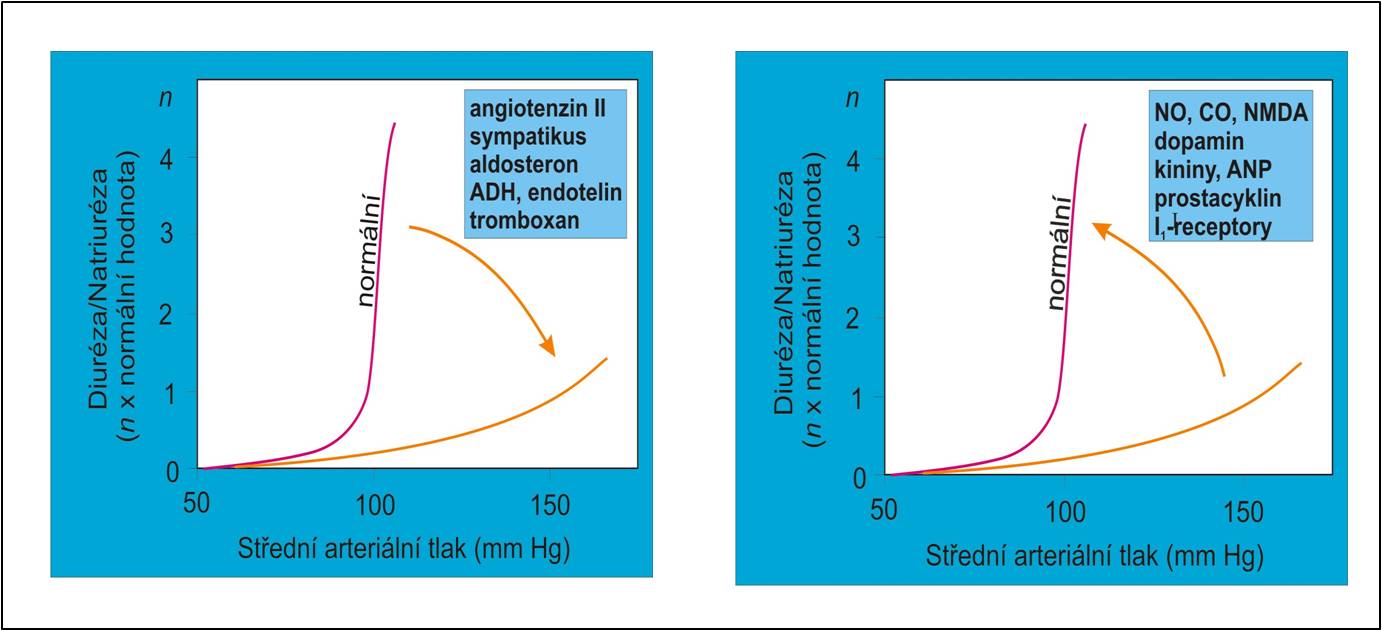
Obr. 3. Humorální faktory podílející se na snížení náklonu (vlevo) a zvýšení náklonu (vpravo) ledvinové funkční křivky. (Podle AC Guyton a JE Hall: Textbook of Medical Physiology, 11th Ed. Elsevier/Saunders, Philadeplhia. 2006.) Schéma je možno zhlédnout i v animované podobě.
Hypertenze volumového typu se skloněnou renální funkční křivkou (primárně neprovázená vazokonstrikcí)
Obecný popis vývoje hypertenze objemového typu
Počáteční změny. Hyperkinetická cirkulace. Iniciální fáze objemové hypertenze se projevuje hyperkinetickou (hyperdynamickou) cirkulací. Jejími průvodními rysy jsou snížený periferní odpor, tachykardie, zvýšený srdeční výdej, a vegetativní labilita.
Nárůst arteriálního tlaku je v iniciální fázi objemové hypertenze způsoben hlavně zvětšením srdečního výdeje na základě vysokého žilního návratu. Vysoký žilní návrat je přímým důsledkem zvětšení volumu cirkulující krve. To je možné jen při poruše ledvin. Charakterizovat tuto fázi jako hyperkinetickou cirkulaci s vysokým srdečním výdejem případně s nedostatečně redukovaným periferním odporem bez zřetele na vysoký žilní návrat, jak to lze dosud najít v některých učebnicích, je nedostatečné. Podstatou problému už v této fázi je nepoměr mezi náplní a kapacitou krevního řečiště (při abnormální funkční křivce ledvin). Představa některých autorů o vysokém srdečním výdeji, který by při nedostatečně redukovaném periferním odporu nebyl podložen zvětšeným efektivním objemem a tlakem krve, je sama o sobě rozporná. Srdce by nemohlo pumpovat velké objemy krve a udržovat vyšší krevní tlak, kdyby nebylo náležitě zásobeno odpovídajícím žilním návratem.
Změny provázející vývoj volumové hypertenze jsou shrnuty na obrázku 4.
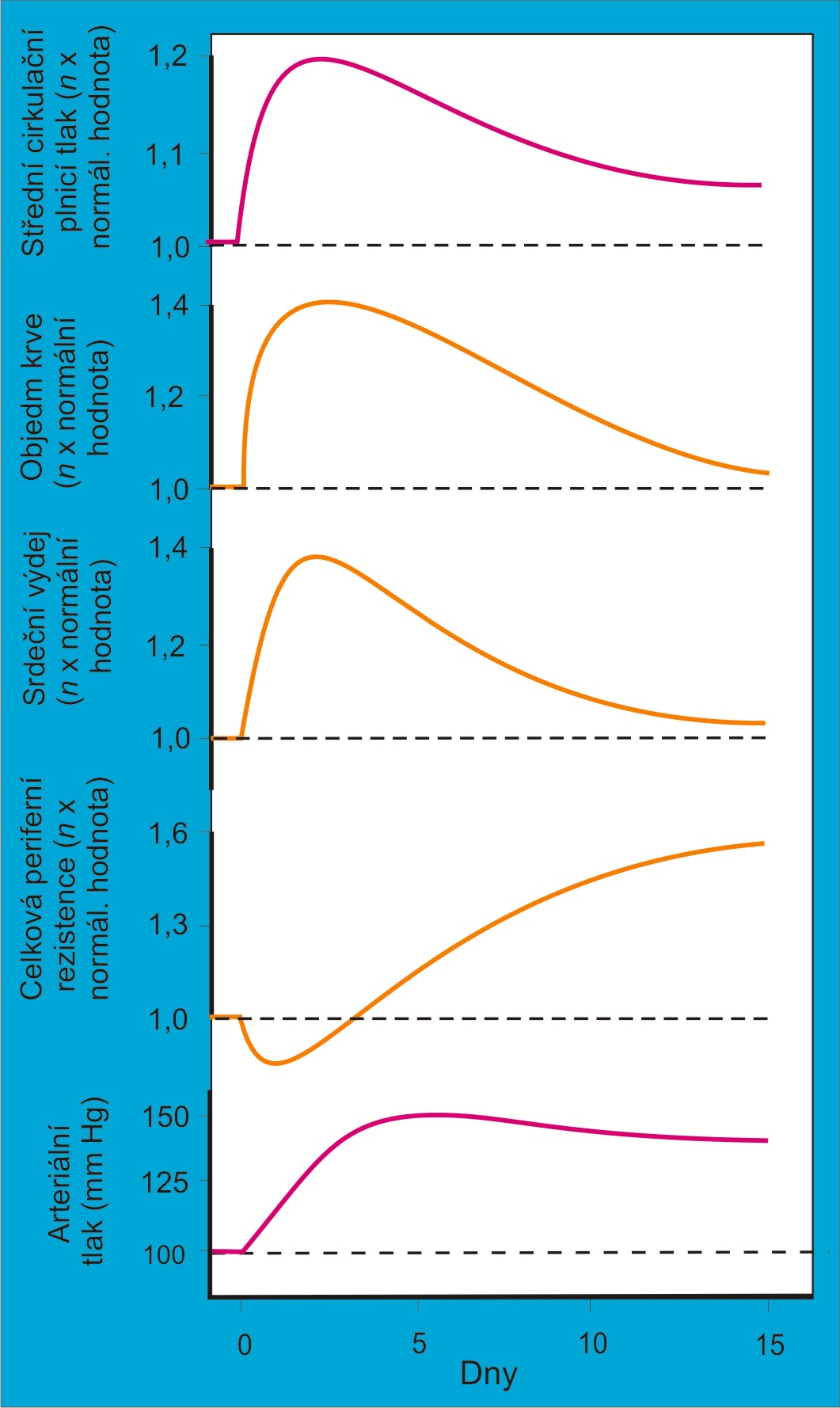
Obr. 4. Změny objemu krve, srdečního výdeje, periferní rezistence, středního plnicího cirkulačního tlaku a arteriálního tlaku od počátku do ustálení objemové hypertenze. (Podle AC Guyton a JE Hall: Textbook of Medical Physiology, 11th Ed. Elsevier/Saunders, Philadeplhia. 2006.)
Změny jednotlivých veličin znázorněné na obrázku 4 jsou následující.
Primární změnou je zvětšení objemu cirkulující krve. Větší objem cirkulující krve zvyšuje střední cirkulační plnicí tlak, následně žilní návrat a srdeční výdej (obr. 4).
Periferní cévní odpor v iniciální fázi vývoje volumové hypertenze v systémové cirkulaci klesá (obr. 4). Tento fakt lze v prvních dnech začínající volumové hypertenze uspokojivě vysvětlit zapojením krátkodobé nervové reflexní kontroly tlaku. Jde o mimořádně významný rys, který znovu ukazuje na dvě slutečnosti:
- Vazokonstrikce nemusí provázet hypertenzi od počátku;
- Vazokonstrikce není primární podmínkou vzniku hypertenze.
Objemová hypertenze vzniká z počátečního nevelkého nadbytku intravaskulární tekutiny udržovaného poruchou ledvin. Už chronické zvětšení krevního objemu o několik procent, trvá-li dostatečně dlouho, vyvolá klinicky významné zvýšení krevního tlaku.
Další vývoj změn periferní rezistence. Úloha lokální tkáňové autoregulace při zvyšování periferního cévního odporu při objemové hypertenzi. Vzhledem ke snížení periferního odporu v iniciální fázi nemusí počáteční nárůst objemu krve příliš zvětšit ani srdeční výdej, ani arteriální tlak. Pokud ale nedojde k jejich včasné nápravě, tzn. k opětovné normalizaci volumu, spustí se plynulý sled kroků, v nichž se souběžně a ve vzájemných závislostech mění arteriální tlak, krevní průtok tkáněmi, periferní odpor a srdeční výdej. Tento proces trvá týdny až měsíce.
Kritickým článkem uvedených změn v následující fázi je zvyšování periferního odporu (obr. 4). Přírůstky odporu postupně násobí počáteční zvýšení arteriálního tlaku (obr. 4). Podnětem ke změnám periferního odporu je příliš vysoký průtok krve tkáněmi, diktovaný zvýšeným arteriálním tlakem. Vysoký průtok vyvolá lokální tkáňovou autoregulační odpověď. Výsledkem této řetězové reakce, která byla nazvána „tlakovým multiplikátorem“, je, že původní, zdánlivě nevýznamné zvětšení objemu o 2 – 3 %, udržované alterovanou funkční ledvinovou křivkou, zvýší tlak až o 30 – 50 %. Změny periferního odporu, které se u volumové hypertenze vyvíjejí až sekundárně (obr. 4), tak jsou zcela pod kontrolou autonomní lokální tkáňové autoregulace průtoku krve.
Postupným řetězcem změn se v srdci rozvíjí hypertrofie. Stěny velkých arterií ztrácejí pružnost jednak proliferací a hypertrofií jejich elementů, jednak ukládáním kolagenu a vaziva. Remodelace zahrnuje intimu, medii i adventicii. Vytváří se dokonce nová lamina elastica interna. Malé cévy nejprve reagují přeskupením vláken hladkého svalstva, aniž zpočátku musí dojít ke ztluštění jejich stěn. Později se ale ani ony nevyhnou hypertrofické remodelaci. Změnám předchází vznik endoteliální dysfunkce. Při hypertenzi se na ní významně podílí zvýšené smykové tření. Konečně při hypertenzi dochází ke snížení hustoty sítě mikrocév. Změna probíhá ve dvou fázích. První fáze je funkční a vratná, neprovázená anatomickými změnami. Mikrocévy jsou přítomny, ale většinou zůstávají uzavřeny a nejsou perfundovány. Za toto uzavření cév není odpovědný sympatikus – denervace nevede k naplnění cév. Druhá fáze je anatomická. Během ní může snížení hustoty arteriol dosáhnout až 25 – 40 % oproti výchozímu stavu.
Výsledkem postupného navyšování periferního odporu je, že počáteční hyperkinetická cirkulace se začíná transformovat do podoby cirkulace se zvýšenou periferní rezistencí. Ustálenou hypertenzi pak už po celé další období provází vysoký periferní odpor. Zvýšený cévní odpor je charakteristickou a konstantní složkou každé rozvinuté (chronické) hypertenze. Z toho se dříve vyvozovalo, že samotná změna periferního odporu je primární příčinou chronického zvýšení arteriálního tlaku. U volumové hypertenze ovšem začíná periferní odpor narůstat až po přechodné fázi, během níž dokonce došlo k jeho poklesu. Zvýšení rezistence následuje, nikoli předchází, hypertenzi. Nemůže být její příčinou, protože krevní tlak byl zvýšen už v období, kdy periferní odpor ještě byl nízký. Růst periferního odporu je sekundární změnou!
Návrat srdečního výdeje k normě. Se zvýšením celkového periferního odporu klesá žilní návrat, a tím i srdeční výdej. Obě tyto veličiny se v další fázi vývoje hypertenze postupně vracejí prakticky k normě (obr. 4). To je důležité pro myokard, protože vysoký srdeční výdej zatěžuje srdce.
Charakteristiké rysy rozvinuté volumové hypertenze. Hypertenzi, která se od počátku vyvíjela jako volumová, pak v její rozvinuté (chronické) podobě charakterizuje téměř normální objem krve a téměř normální srdeční výdej. Kromě zvýšeného arteriálního tlaku přetrvává výrazně zvýšený periferní odpor a střední cirkulační plnicí tlak (obr. 4). Hemodynamické poměry u pacientů s rozvinutou volumovou hypertenzí se tak diametrálně liší od poměrů popsaných u začínající volumové hypertenze.
Krátkodobé a dlouhodobé mechanismy úpravy periferního odporu. Na popsaném zvýšení periferní rezistence, které se rozhodujícím způsobem podílí na přechodu volumové hypertenze do chronického stadia, se podílejí krátkodobé a dlouhodobé mechanismy:
- Cévy hypertenzivních jedinců jsou zvýšeně vnímavé na vazopresorické a méně citlivé na vazodepresorické podněty. Myogenní vazokonstrikční odpověď, založená na přirozené reakci hladkých svalů na zvýšený tlak, dosahuje u hypertenzivních jedinců mnohem větší intenzity.
- V dlouhodobém průběhu hypertenzi provází nevratná hypertrofická přestavba stěn arterií, která zmenšuje jejich průsvit. Navíc dochází k prokazatelnému prořídnutí mikrocirkulace, které je ověřitelné měřením hustoty kapilár v sítnici, spojivce anebo nehtovém lůžku. Prořídnutí mikrocirkulace bylo následně histologicky potvrzeno i ve vzorcích kosterního svastva, srdečního svalstva a žaludku.
Vývoj patologických změn v ledvinách při objemové hypertenzi. Glomerulární hypertenze a hyperfiltrace, která provází objemovou hypertenzi, poškozuje ledvinové cévy a glomeruly jednak mechanicky, vyšším pnutím stěn a smykovým třením proudící tekutiny, jednak větším objemem filtrátu a množstvím proteinů, které do něho pronikají. Aktivují se mesangiální buňky, které jsou vybaveny stažlivými elementy a mají schopnost fagocytózy. Zvyšuje se produkce cytokinů a růstových faktorů. S postupem hypertenzní nemoci se vyvíjí arterioloskleróza a glomeruloskleróza a glomerulární filtrace klesá. V období, kdy se manifestuje rozvinutá hypertenze, už glomerulární filtrace může být snížena pod normální hodnoty a může být provázena morfologickou alterací glomerulů. Glomeruloskleróza vyvolaná hypertenzí je po diabetické nefropatii druhou nejčastější příčinou chronického selhání ledvin.
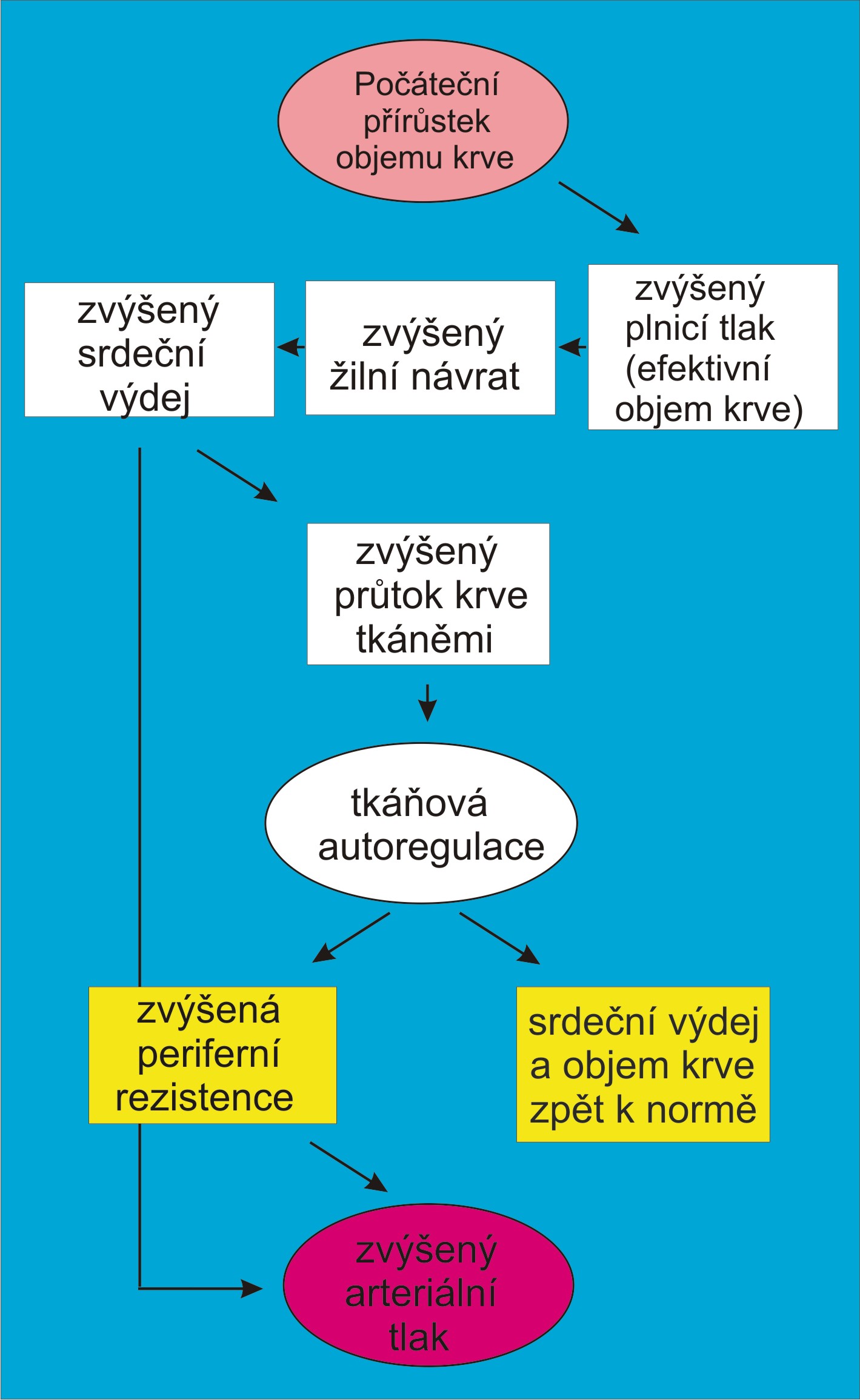
Obr. 5. Schematické znázornění vývoje objemové hypertenze.
Ustálení hypertenze zaručuje, že ledviny dlouhodobě budou mít dostatečný perfúzní tlak, aby vyloučily potřebné množství iontů a tekutin a zabezpečily vyrovnanou bilanci. Jedinci s hypertenzí vylučují stejná množství soli a vody jako zdraví; ledviny zdravých osob by ovšem při stejném tlaku vylučovaly 2 – 4 x více tekutin než ledviny hypertenzivních pacientů. Díky zvýšení tlaku se nehromadí zplodiny metabolismu a nejsou zjevné žádné známky porušených ledvinových funkcí. To vede klinické pracovníky k závěru, že ledviny jsou v pořádku. Ledviny však mají abnormální funkční křivku. Dosud nebyla popsána žádná hypertenze, u níž by funkční křivka ledvin byla beze změny. Insuficience ledvin se klinicky projeví po snížení krevního tlaku.
Terapeutické souvislosti. Je namístě věnovat pozornost tomu, že při úspěšném terapeutickém použití diuretik se krevní tlak nejprve snižuje redukcí přílišného krevního volumu, žilního návratu a srdečního výdeje. Toto vysvětlení je alternativou k pravděpodobně nesprávné představě, že za antihypertenzivní účinek moderních diuretik odpovídají jejich vazodilatační účinky. Úplná úprava tlaku závisí na tom, nakolik se podaří trvale ovlivnit ledvinovou funkční křivku. Dalším obtížným problémem terapie hypertenze je náprava periferní rezistence. Změny jsou tím méně vratné, čím delší bylo trvání hypertenze a čím byla těžší. Je vhodné si také všimnout, že expanze extracelulárních tekutin u volumové hypertenze inhibuje renin-angiotenzinový systém. Tento typ hypertenze tedy přinejmenším v časných stadiích nemusí reagovat na inhibitory ACE anebo angiotenzinových receptorů. Situace se však může změnit, pokud v dalším vývoji dojde k sekundárním arteriolopatiím a glomerulopatiím. Klinické údaje nasvědčují, že podávání inhibitorů může zabrzdit vývoj sekundárních změn.
Příklady hypertenzí objemového typu
Mineralokortikoidní hypertenze. Primární hyperaldosteronismus.
Nadbytek mineralokortikoidů produkovaných adenomem (Connův syndrom) aktivuje epitelový natriový kanál (ENaC) na luminálních membránách distálních tubulů. Mineralokortikoidy zároveň stimulují transportní Na+/K+-ATPázu na bazolaterální membráně, která přečerpává ionty Na+ do intersticia ledvin. Tyto procesy zmenšují sklon renální funkční křivky. Zvýšené koncentrace aldosteronu v plazmě způsobí v prvních několika dnech retenci iontů sodíku. Zásoby sodných iontů v těle se v této fázi zvětší a zůstanou už zvětšené přinejmenším tak dlouho, dokud trvá hypersekrece aldosteronu. Výsledkem tohoto období, provázeného negativní bilancí sodných iontů, je expanze objemu extracelulární tekutiny a nárůst středního cirkulačního plnicího tlaku. Zvětšuje se žilní návrat a srdeční výdej. Vyvíjí se typická objemová hypertenze citlivá k množství soli přiváděné s potravou (obr. 4 a 5).
Stupeň hypertenze u primárního hyperaldosteronismu je určen výší tlaku, při níž tlaková natriuréza nakonec právě vyrovná zpětnou resorpci iontů stimulovanou hormonem (obr. 6). Natriuréza se v průběhu několika dnů znovu vrátí k normě. Ustálí se nový rovnovážný stav. Hospodaření sodných iontů znovu dosáhne vyrovnané bilance. Sodné ionty se potom už v těle dále neakumulují. Dochází k úniku vylučování Na+ iontů zpod reabsorpčního působení aldosteronu. Tento jev se nazývá „fenomén úniku“ („escape phenomena“) (obr. 6). Vzhledem k uplatnění fenoménu úniku se sysstémový arteriální krevní tlak u primárních mineralokortikoidních hypertenzí příliš nezvyšuje. Ani objem plazmy příliš neroste, a rovněž srdeční výdej bývá jen mírně zvýšený. Tlaková diuréza nakonec neutralizuje zvýšenou reabsorpci Na+, podmíněnou přítomností aldosteronu, a expanze extracelulárních tekutin a zvyšování krevního tlaku dosáhne limitu úměrného koncentraci aldosteronu v plazmě. Dokud hladina aldosteronu zůstává vyšší, musí zůstat zásoba sodných iontů vtěle a arteriální tlak zvýšené.
Obr. 6. Účinek aldosteronu a jiných sůl- a vodu-šetřících hormonů na ledvinovou funkční křivku a fenomén úniku. (Podle Hall JE: Am. J. Physiol. 277 (Adv. Physiol. Educ. 22), S174-S186, 1999.) Schéma je možno zhlédnout i v animované podobě.
Systémová arteriální hypertenze a fenomén úniku se objevuje při hypersekreci všech natrium- anebo vodu-šetřících hormonů. Nejde tedy jen o mineralokortikoidy, ale také o glukokortikoidy, angiotenzin II, antidiuretický hormon, katecholaminy atd. Zasluhuje zvláštní pozornost, že po několikaměsíčním trvání už ani operativní odstranění tumoru (zrušení hypersekrece) nemusí vést k úpravě cévní rezistence a krevního tlaku zpátky na normu.
Zajímavým a do jisté míry i paradoxním rysem chronického primárního hyperaldosteronismu je polyurie. Polyurie je způsobena poruchou koncentrační schopnosti tubulů při hypokalémii, která chronický aldosteronismus provází. Na druhé straně je přítomna hypernatrémie, která vede k polydipsii a kompenzuje ztráty tekutin.
Primární zvýšená sekrece antidiuretického hormonu – syndrom nadměrné sekrece antidiuretického hormonu
Antidiuretický hormon (ADH) kontroluje tzv. fakultativní zpětné vstřebávání vody v distálních částech nefronu. Jde o asi 10 – 20 % z celkového objemu glomerulárního filtrátu, tedy o zbytek po tzv. obligátní reabsorpci tekutin v proximálních částech nefronů. Těchto 10 – 20 % představuje kolem 20 – 30 l vody za den; pacienti s deficitem sekrece ADH mohou toto množství denně ztrácet.
Nadměrná sekrece ADH způsobuje zvýšení zpětné reabsorpci vody. Následně se zvětšuje objem extracelulární tekutiny a tekutiny v cirkulaci (obr. 4 – 6). Jako u všech objemových hypertenzí se může i u tohoto syndromu objem zvětšovat jen do doby, než nárůst arteriálního tlaku a následná tlaková diuréza převáží resorpční kapacitu tubulů stimulovanou hormonem („fenomén úniku„) (obr. 6). Vzestup objemu proto nebývá velký. Ledviny stimulované ADH šetří vodu ale vylučují koncentrovanou moč s velkým množstvím sodných iontů. Osmolalita moči bývá dokonce vyšší než osmolalita plazmy. Naproti tomu koncentrace Na+ v extracelulární tekutině je silně snížená. Těžký deficit iontů sodíku může vést až k příznakům otravy vodou (křeče a koma). Kromě ADH se na deficitu osmolality podílí i potlačení sekrece reninu a aldosteronu hypervolémií.
Při syndromu hypersekrece ADH je vcelku mnohem více postiženo složení tělních tekutin než jejich objem. To výmluvně dokumentuje, že ADH není hormonem kontroly objemu, nýbrž regulátorem koncentrace iontů Na+. Hypotalamické receptory koncentrace sodných iontů jsou při syndromu nadměrné sekrece ADH nastaveny na nižší cílovou hodnotu osmolality. Vysoké hladiny hormonu jsou pak přítomny i přes život ohrožující hyponatrémii.
Molekulární hypertenze
Molekulární formy hypertenze se donedávna řadily do skupiny esenciálních hypertenzí. Dnes je jasné, že se jedná o hypertenze volumového typu provázenou charakteristickou volumovou poruchou funkční křivky ledvin. Jsou souhrnně ilustrovány na obr. 7.
Obr. 7. Molekulární formy systémové arteriální hypertenze. Hyperaldosteronismu a pseudohyperaldosteronismus ve vývoji objemové hypertenze. L, luminální strana; I, strana epitelové buňky přivrácena do intersticia. Ostatní zkratky jsou vysvětleny v textu. (Převzato se svolením autora a nakladatele z J Veselý: Tlaková diuréza a arteriální hypertenze. Epava, Olomouc, 2002.)
Kongenitální adrenální hyperplázie provázená hypertenzí
Hypertenze provází asi 10 % případů vzácné kongenitální adrenální hyperplázie. Jsou z největší části způsobeny autozomální recesívní poruchou 11β-hydroxylázy a z menší části autozomální recesívní poruchou 17α-hydroxylázy. Tyto poruchy se vyznačují neschopností syntetizovat kortizol. V nadměrné míře se uvolňují mineralokortikoidní steroidy (obr. 7).
Zbývajících asi 90 % případů adrenální hyperplázie je způsobeno defektem 21-hydroxylázy, enzymu potřebného pro syntézu jak glukokortikoidů, tak mineralokortikoidů. Provází je hypotenze.
Hyperaldosteronismus příznivě ovlivnitelný glukokortikoidy
Tato autozomálně dominantní porucha má vysokou penetranci a je způsobena nesymetrickou rekombinací genů na lidském chromozomu 8. Rekombinace napojuje jeden z klíčových genů tvorby aldosteronu, gen aldosteronsyntázy (18-hydroxylázy), na regulační sekvenci genu 11β-hydroxylázy, čímž převádí sekreci aldosteronu pod kontrolu ACTH. Aldosteron se pak ektopicky produkuje v zona fasciculata. Výsledkem je nadprodukce aldosteronu (obr. 7) řízená ACTH a odtud odvozená sůl-senzitivní hypertenze provázená nízkou plazmatickou koncentrací reninu a angiotenzinu II. Podle očekávání jsou přítomny hypokalémie a vysoké ztráty K+ iontů močí. Stav lze zmírnit podáním glukokortikoidů, které sníží hladinu ACTH.
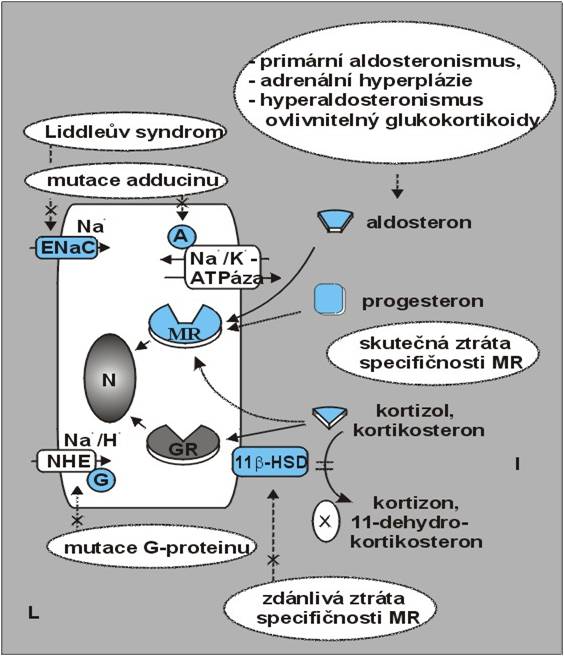
Zdánlivá ztráta specifičnosti mineralokortikoidních receptorů – pseudohyperaldosteronismus způsobený defektem 11β-hydroxysteroiddehydrogenázy
Mineralokortikoidní receptor je nitrobuněčný cytoplazmatický protein, který vedle aldosteronu může vázat i glukokortikoidní hormon kortizol. Kortizol cirkuluje v krvi v koncentracích o několik řádů vyšších než aldosteron. Buňky, které selektivně reagují na aldosteron, mají na svém povrchu enzym 11β-hydroxysteroiddehydrogenázu (obr. 7). Ta účinně mění kortizol v neaktivní kortizon a zajišťuje, že v okolí těchto buněk je lokální přebytek aldosteronu.
Autozomálně recesívní defekt genu 11β-hydroxysteroiddehydrogenázy na chromozomu 11 vede k nedostatečné inaktivaci kortizolu. Kortizol proniká do buněk a interaguje s receptory aldosteronu. Defekt se manifestuje jako zdánlivý nadbytek mineralokortikoidů v distálním tubulu a jako zdánlivá ztráta specifičnosti mineralokortikoidních receptorů. Výsledkem je hromadění iontů Na+ a vody v organismu a sůl-senzitivní volumová hypertenze. Rovněž hypertenze po otravě extrakty z lékořice je pravděpodobně způsobena hlavně inhibicí 11β-hydroxysteroid dehydrogenázy. Podobný obraz má hypertenze z nadprodukce kortizolu provázející Cushingův syndrom anebo defekty glukokortikoidních receptorů.
V protikladu k předchozí poruše glukokortikoidy tento defekt zhoršují. Příznivě naproti tomu působí blokáda mineralokortikoidních receptorů spironolaktonem.
Skutečná ztráta specifičnosti mineralokortikoidních receptorů – molekulární porucha mineralokortikoidního receptoru jako příčina preeklampsie
Fyziologickou odpovědí v těhotenství je hyperkinetická cirkulace z nově otevřených krevních cest v placentě provázená mírným poklesem krevního tlaku. Asi v 1 – 6 % případů se u těhotných žen vyvíjí hypertenze. Hypertenze zvyšuje riziko preeklampsie, která ohrožuje matku i plod. Preeklampsie se obvykle manifestuje po 20. týdnu těhotenství. Lze předpokládat, že se do té doby vyvíjí latentně. Pokud se k hypertenzi přidá proteinurie, pak se bez ohledu na rozsah proteinurie nedá osud pacientky spolehlivě předvídat a může rychle progredovat do konvulzivní formy eklampsie.
S preeklampsií souvisí jeden z nejnovějších fascinujících objevů na poli sůl-senzitivní hypertenze. U těžké hypertenze vzniklé během těhotenství byla zjištěna vzácná mutace mineralokortikoidního receptoru, která spočívá v záměně serinu v pozici 810 vazebné domény receptoru za leucin. Mutace mění afinitu receptoru ke steroidům (obr. 7). Mutovaný receptor má vysokou afinitu k progesteronu. Fyziologický vzestup hladiny progesteronu v průběhu těhotenství vede k obsazení mineralokortikoidního receptoru progesteronem, stimulaci receptoru a k expresi jím řízených genů. Výsledkem je akumulace Na+ a tekutin a těžká hypertenze. Spironolakton se chová jako agonista mutovaného receptoru, a je proto přísně kontraindikován. Vzhledem k tomu, že mineralokortikoidní receptor s bodovou mutací Ser810Leu je konstitutivně aktivní, nebo může být stimulován dalšími steroidy, může se hypertenze vyskytovat i u příbuzných jedinců mužského pohlaví anebo u negravidích příbuzných žen.
Pro úplnost je namístě upozornit, že uvedená vzácná mutace vysvětluje jen nepatrný zlomek případů preeklampsie. Kromě vysokého arteriálního tlaku jsou pro preeklampsii charakteristické:
- Zvýšená periferní rezistece a vazokonstrikce s vysokou citlivostí cév na vazopresorické podněty, zejména na angiotenzin II a katecholaminy. Angiotenzin II se ve zvýšené míře váže i na krevní destičky, alespoň u části nemocných, což je možno využít diagnosticky.
- Snížený krevní objem se sníženým srdečním výdejem.
- Proteinurie a otoky, poruchy koagulace a poruchy jaterních funkcí.
Patologicko-anatomickým nálezem typicky provázejícím preeklampsii je zbobtnání buněk glomerulů a glomerulárních kapilár („glomerulární endotelióza“). Na rozdíl od právě uvedené molekulární formy preeklampsie bývají v počátečních stadiích preeklampie často přítomny vysoké hladiny reninu a angiotenzinu II, přinejmenším do doby, než arteriální tlak dosáhne výše potřebné k vyrovnané bilanci tekutin. Potom může reninová aktivita klesat. Bývají zvýšeny hladiny natriuretických atriopetidů.
Omezená funkce glomerulů řadí preeklampsii do blízkosti stavů charakterizovaných úbytkem funkční ledvinové masy. Jejich společným rysem je postižení filtračního koeficientu. Toto hledisko podporuje i mimořádná citlivost této hypertenze k příjmu soli. Vzhledem k vysokému cévnímu odporu a nízkému krevnímu objemu by však bylo možno preeklampsii přiřadit i k hypertenzím vazokonstrikčního typu.
Až na souvislost s těhotenstvím připomíná fenotyp poruchy Ser810Leu fenotyp jiného onemocnění, tzv. Liddleova syndromu popsaného níže. Kromě hypertenze je zde rovněž přítomna hypokalémie, je potlačena aktivita reninu a inhibována sekrece aldosteronu; lze připomenout, že i Liddleův syndrom je refrakterní na spironolakton.
Poruchy transportních systémů pro Na+ v ledvinových tubulech
Primární hybné gradienty, které umožňují reabsorpci Na+ v ledvinách, vytváří transportní Na+/K+-ATPáza přítomná na bazolaterálních membránách tubulárních buněk. Zvýšená aktivita tohoto systému může být odpovědná za pokles směrnice ledvinové funkční křivky, akumulaci iontů sodíku a tekutin v organismu a následně za hypertenzi objemového typu. K takové činnosti může být Na+/K+-ATPáza neadekvátně stimulována řadou přímých i nepřímých podnětů.
Epiteliální Na+-kanál ENaC: Liddleův syndrom – pseudohyperaldosteronismus způsobený mutací β- nebo γ-podjednotky ENaC
Jedna z poruch, při nichž se zvyšuje koncentrace iontů Na+ v tubulárních buňkách, kde stimulují Na+/K+-ATPázu, je způsobena změnou struktury epitelového Na+ kanálu (ENaC) regulovaného aldosteronem a citlivého k amiloridu. Jde o aktivující mutaci neboli mutaci typu „gain-of-function“ ENaC. Nazývá se Liddleův syndrom (obr. 7).
Kanál ENaC je v ledvinách umístěn na apikálních membránách obrácených do lumina distálních částí tubulů. Je složen ze tří podjednotek, α, β a γ. Mutace, které způsobují Liddleův syndrom, jsou lokalizovány v genech pro strukturní podjednotky β a γ na lidském chromozomu 16. Mutované kanálové jednotky nejsou schopny dostatečně rychlé internalizace a setrvávají na apikálních membránách i po vymizení náležitého podnětu (např. po snížení koncentrace aldosteronu). Pokud je kanál takto pozměněn, zůstává konstitutivně aktivní a umožňuje transport velkého množství sodných iontů. Funkční křivka ledvin nemůže dosáhnout normální strmosti. Antagonista aldosteronu spironolakton s mutovaným receptorem neinteraguje, a je proto neúčinný. Naopak, může ho dokonce stimulovat. (Alternativní inaktivující mutace neboli mutace typu „loss-of-function“ kanálu ENaC nezpůsobují hypertenzi, nýbrž hypotenzi s hyperkalémií a acidózou (pseudohypoaldosteronismus).)
Sekrece reninu je u Liddleova syndromu utlumena hypertenzí a zvýšeným obsahem Na+ a tekutin v těle. Rovněž hladiny aldosteronu jsou minimální, což ospravedlňuje termín pseudohyperaldosteronismus. Aldosteron se nevyplavuje ani po drastickém omezení příjmu NaCl. Tento jev lze vysvětlit inhibicí aldosteronsyntázy (18-hydroxylázy) v nadledvinách chronickou hypervolémií. Elektrogenní ENaC spoluvytváří elektrický hybný gradient pro sekreci iontů K+ a H+. Proto je u Liddleova syndromu častá hypokalémie, která ale nebývá přítomna vždy, a alkalóza.
Porucha výměnného transportního systému Na+/H+
Zvýšená aktivita Na+/H+-antiportéru (NHE) je klasickým znakem genetické predispozice k hypertenzi. Vyskytuje se u velké části (40 – 50 %) hypertenzivních pacientů i u zatím zdravých potomků hypertenzivních rodičů. Fenotypově se projevuje i mimo ledviny. Z toho důvodu je možné jeho testování na erytrocytech, a to buď přímo, nebo jako výměnu Na+/Li+. Za vysokou aktivitu Na+/H+-výměnného systému v ledvinách hypertenzivních osob není odpovědná ani nadměrná exprese NHE, ani jeho mutace, nýbrž genetický polymorfismus podjednotky jednoho z G-proteinů, se kterým NHE interaguje (obr. 7). Podjednotka Gβ3 trimerního G-proteinu má v pozici 825 zaměněn cystein za threonin.
Defekty cytoplazmatických složek transportních systémů – abnormální stimulace Na+/K+-ATPázy mutovaným adducinem
ATPáza je ovlivňována nejen kinetickými faktory, jako jsou změny koncentrací iontů Na+ a K+ uvnitř anebo vně tubulárních buněk, ale také přímými strukturními interakcemi se složkami buněčné membrány a jejího nejbližšího okolí. Jednou z takových složek je heterodimerní cytoskeletární protein adducin (obr. 7). Adducin interaguje s aktinem, účastní se procesů jeho polymerace, zprostředkuje tvorbu komplexu mezi aktinem a spektrinem a zasahuje do činnosti signalizačních kaskád v buňce, zejména pokud jde o transport iontů. Předpokládá se, že se podílí na přesunech Na+/K+-ATPázy mezi povrchem buňky a cytoplazmou při zvyšování anebo snižování její hustoty v bazolaterálním úseku buněčné membrány (senzitizace a desenzitizace systému).
Adducin je složen z podjednotek α a β. Pacienti, kteří mají ve svém genotypu „hypertenzivní“ (460Trp-) alelu α-podjednotky adducinu, jsou citliví na příjem soli. Mutovaná varianta zvyšuje reabsorpci iontů sodíku v ledvinách (stimuluje Na+/K+-ATPázu. Kromě toho narušuje přestavbu cytoskeletu vytvářeného aktinovými vlákny, takže může ovlivňovat i jiné Na+-dependentní transportní systémy. Následkem je zmenšení sklonu ledvinové funkční křivky a vystupňování reabsorpčních dějů. Nositelé 460Trp-alely dosahují nižších hodnot tlakové natriurézy a diurézy. Mají nízkou plazmatickou reninovou aktivitu. Tito pacienti dobře reagují na diuretickou léčbu.
Defekty cytoplazmatických regulačních složek transportních systémů – mutace serin/treoninových proteinkináz WNK
Jiné familiální defekty, které vedou k pseudoaldosteronismu, spočívají v aktivačních mutacích serin/treoninových kináz skupiny WNK. Odpovídající geny byly lokalizovány na chromozomech č. 12 (WNK1) a 16 (WNK4). Obě proteinkinázy se vyskytují v buňkách distálních částí nefronu, které jsou pod kontrolou aldosteronu. Zatímco WNK1 je v cytoplazmě, WNK4 je jak v cytoplazmě, tak v intercelulárních spojeních neprůlinčitého typu. Defekty WNK jsou charakterizovány zvýšenou reabsorpcí Na+, hypertenzí, hyperkalémií a hyperchloremickou acidózou.
Funkce proteinkináz WNK zatím nejsou do detailu známé. Pokud mutovaná forma WNK4 zvyšuje průlinčitost mezibuněčných spojení v distálních segmentech, mohlo by to vést k paracelulární resorpci Clˉ iontů a zmenšení elektrického gradientu nutného pro sekreci K+ a H+ iontů v distálních tubulech. Tento scénář by byl v souladu s přítomností hyperkalémie a acidózy.
Poruchy peritubulární dřeňové hemodynamiky a tlaku
Vysoká zpětná tubulární reabsorpce nemusí být způsobena jen funkčními poruchami výstelky ledvinových tubulů, nýbrž také změnami fyzikálních sil v mezitubulárním intersticiu. Zúžení průsvitu a snížení hydrostatického tlaku v postglomerulárním oddílu cirkulace snižuje intersticiální tlak a zvyšuje reabsorpci iontů a tekutin ve dřeni ledvin. Reabsorpci může napomáhat zvýšení onkotického tlaku v eferentních arteriolách a v jejich povodí. Všechny tyto procesy mohou alterovat renální funkční křivku. Zúžení vas efferens může být funkčního, nebo i organického rázu. Změny mohou být obtížně diagnostikovatelné. Jindy pravděpodobně může dojít k nárůstu peritubulárního tlaku a kompresi tubulů, jejichž průchodnost tím trpí. V ledvinách obézních osob např. bylo zaregistrováno zmnožení dřeňových elementů a intersticiální hmoty.
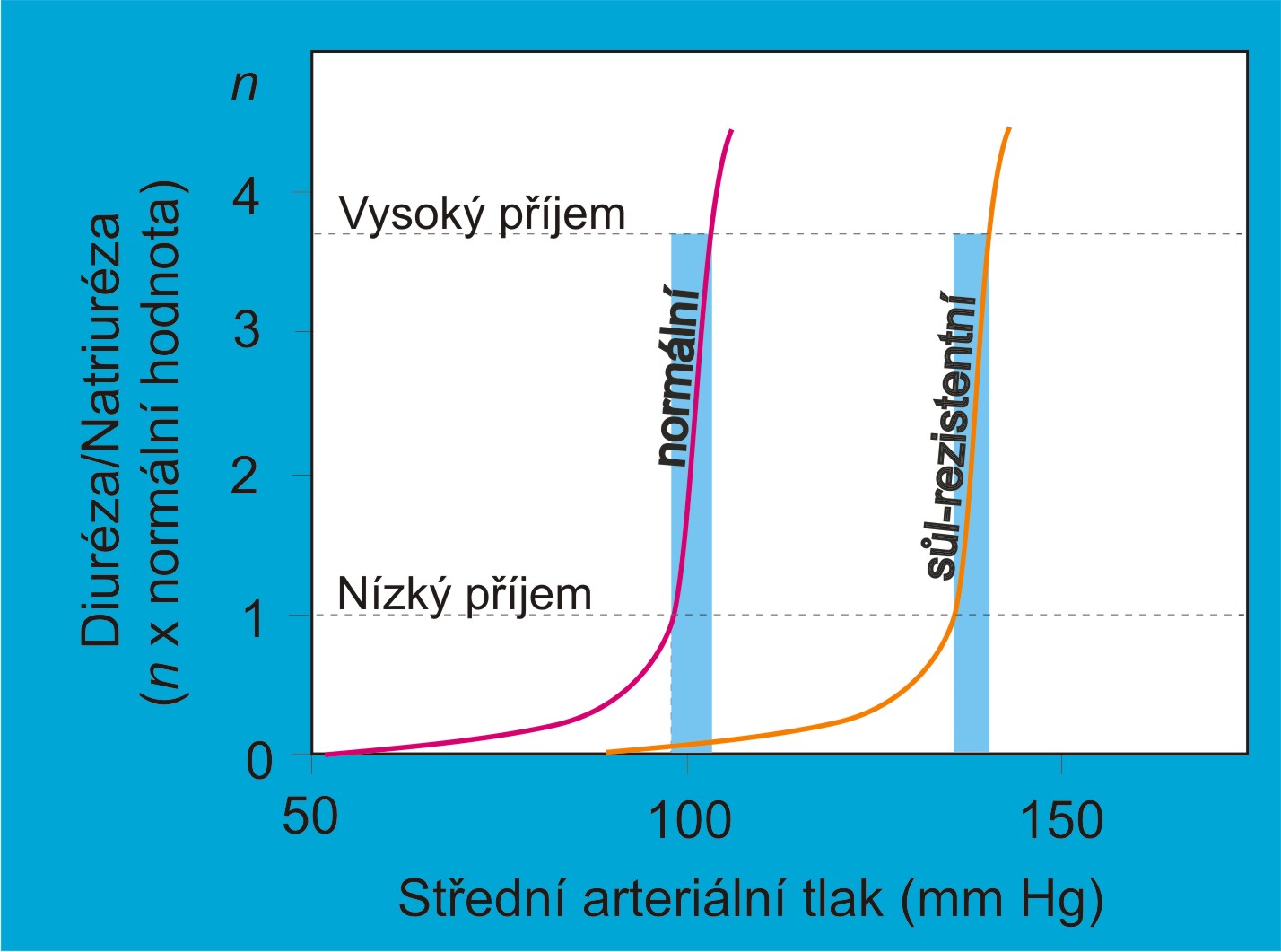
Sůl-senzitivní a sůl-nesenzitivní hypertenze
Část normotenzivních a hypertenzivních osob v populaci reaguje na déletrvající změnu příjmu NaCl změnou systémového arteriálního tlaku. Tyto osoby se označují jako sůl-senzitivní. Pro praktické účely definoval F. Bartter a jeho spolupracovníci citlivost k soli jako nejméně 10% přírůstek středního arteriálního tlaku po změně velikosti příjmu soli. Všechny sůl-senzitivní hypertenze charakterizuje ledvinová funkční křivka skloněná k vyšším hodnotám systémového arteriálního tlaku, která se typisky spojuje s volumovou hypertenzí. Mechanismy odpovědné za citlivost arteriálního tlaku k soli zatím nejsou zcela objasněny. Jde však o závažnou otázku, neboť její řešení by mohlo ukázat, kteří jedinci budou mít prospěch z omezení příjmu soli a kteří pravděpodobně ne. Citlivost k soli je po obezitě druhým nejvýznamnějším nezávislým rizikovým faktorem vzniku hypertenze.
Jednu z cest ke standardizaci klinických vyšetření citlivosti k soli ukazuje pracovní protokol G. Kimury a jeho spolupracovníků. Podle tohoto protokolu dochází ke změnám vysokosolné a nízkosolné diety po týdnu, přičemž volba pořadí je náhodná. Měření tlaku a výdeje sodných iontů se provádějí poslední tři dny v každém týdnu. Tzv. index citlivosti k Na+ je reciproční hodnotou směrnice t odečtené z linearizované tlakově-diuretické (-natriuretické) křivky. Lze ho určit graficky anebo vypočítat vydělením přírůstku středního arteriálního tlaku přírůstkem natriurézy po změně příjmového režimu NaCl (obr. 8).
Index definovaný pomocí reciproční hodnoty směrnice kombinuje několik výhod. Dovoluje kvantifikovat citlivost k soli spojitou stupnicí hodnot při zachování možnosti hodnotit citlivost individuálně, tedy u každého jedince zvlášť. Definuje citlivost k soli nezávisle na velikosti změny jejího příjmu a bez ohledu na to, jestli se vycházelo ze zvýšení nebo snížení příjmu. Pozornost zasluhuje možnost využít pro vyšetření indexu ambulantní 24-hodinové monitorování, aplikované některými skupinami.
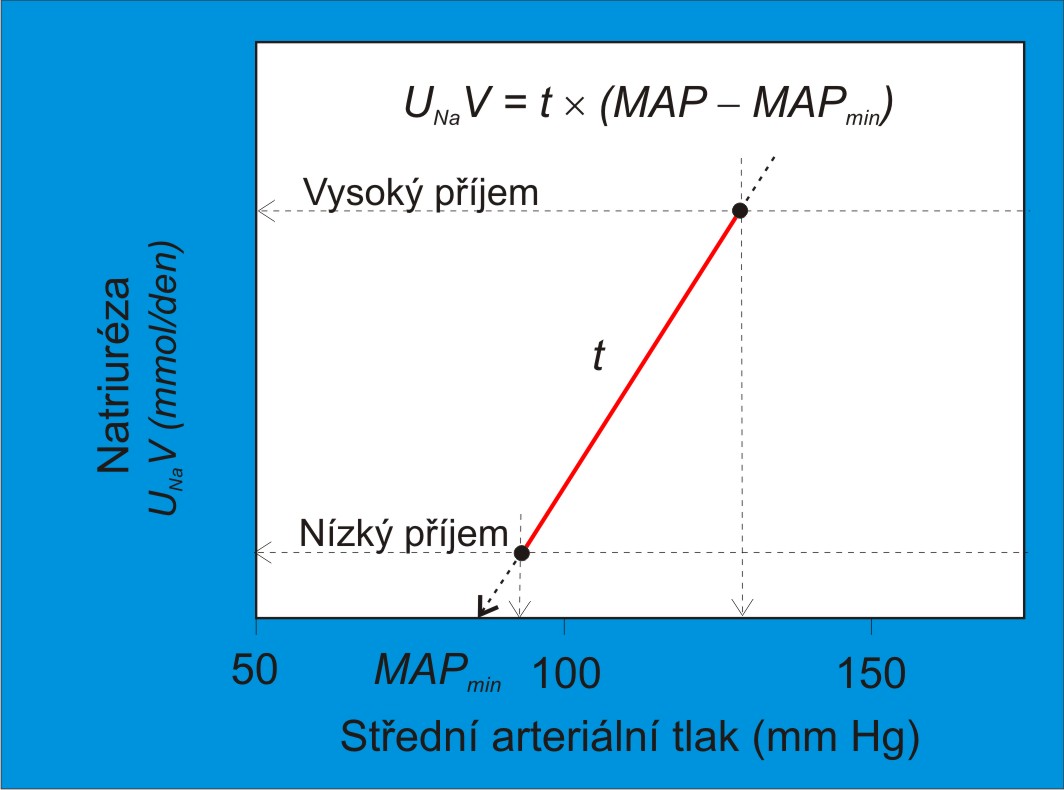
Obr. 8. Ilustrace vyšetření citlivosti ledvinové křivky k soli. (Podle Kimura G, Brenner BM: Curr. Opin. Nephrol. Hypertens. 2, 341-349, 1993.) MAP, střední arteriální tlak; MAPmin, průsečík směrnice s osou x, minimální tlak, při kterém ledvina ještě vylučuje Na+; MAP – MAPmin, efektivní glomerulární filtrační tlak; t, směrnice linearizované funkční ledvinové křivky, její reciproční hodnota je index citlivosti k Na+; UNa+, koncentrace Na+ v definitivní moči; V, objem moče za 24 hodin; UNa+V, denní výdej Na+ močí - rozdíl mezi množstvím Na+ filtrovaným do primární moči a množstvím Na+ reabsorbovaným v tubulech za 24 hodin.
Strmost ledvinové křivky t je rovna UNa+V/(MAP – MAPmin) (obr. 8). Na ní závisí, jak velký bude úhrnný denní výdej Na+ v definitivní moči při tlaku MAP. Sama je určena rozdílem mezi dvěma procesy:
- Glomerulárními, které závisejí na koeficientu glomerulární filtrace, Kf = GFR/(MAP – MAPmin). Bude popsán níže.
- Tubulárními, které reprezentuje tubulární reabsorpce sodných iontů, tNa+, tedy rozdíl mezi množstvím Na+ obsaženým v glomerulárním filtrátu a výdejem Na+ v definitivní moči.
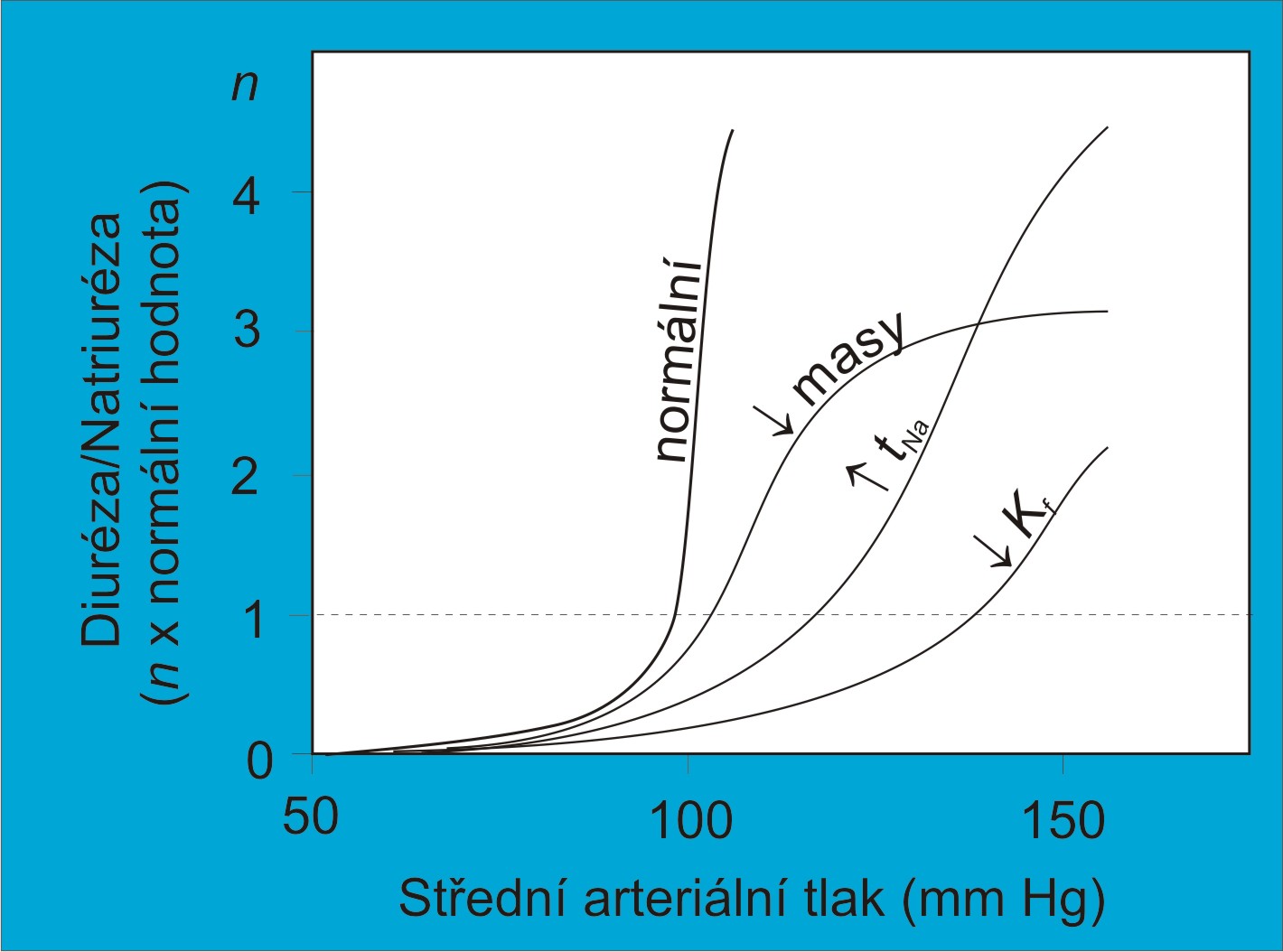
Obecně jsou citlivé k soli všechny stavy se sníženou strmostí funkční ledvinové křivky (obr. 9).
Obr. 9. Variace ledvinové funkční křivky odpovědné za různé formy objemové hypertenze. (Podle AC Guyton a JE Hall: Textbook of Medical Physiology, 11th Ed. Elsevier/Saunders, Philadeplhia. 2006.)
Sůl-senzitivní hypertenze z poruch tubulární reabsorpce
Příklady nadměrné tubulární reabsorpce Na+ byĺy uvedeny výše. Problematika poruch resorpce Na+ však je širší. V evoluci živošišné říše se vazby mezi systémovým arteriálním tlakem, perfúzním tlakem v ledvinách a exkrecí Na+ ustavily asi před 300 – 400 miliony lety. Antinatriuretické hormony noradrenalin, angiotenzin II a aldosteron se začaly výrazněji uplatňovat teprve s přechodem forem života na souš (u obojživelníků a výše). Fylogeneticky staré natriuretické hormony jako dopamin a atriální natriuretický faktor hrály prim ve slanovodním prostředí a dosud převládají u ryb. U suchozemských forem života ustoupily do pozadí, nicméně si podržely svou nezastupitelnou úlohu v regulaci vyrovnané bilance solí a tekutin. Při vzpřímení těla došlo k dalšímu posílení role natrium-šetřících mechanismů. Dnes je zřejmé, že fylogeneticky mladší antinatriuretické mechanismy někdy neúměrně převažují nad svými fylogeneticky staršími natriuretickými protějšky. Jednou za to mohou příliš intenzivní natriumretenční účinky (obr. 3, vlevo), jindy je deficit exkrece iontů Na+ způsoben nedostatečností mechanismů podporujících jejich vylučování (obr. 3, vpravo).
Sůl-senzitivní hypertenze z poruch koeficientu filtrace
Filtrační koeficient Kf je součinem hydraulické permeability, plochy jednotlivého glomerulu a celkového počtu glomerulů. Určuje účinnost glomerulárního tlakového gradientu při tvorbě primární moče (podle rovnice GFR = [MAP – MAPmin] x Kf). Glomerulární postižení, které se patofyziologicky manifestuje nízkým filtračním koeficientem Kf , může spočívat:
- V omezení počtu glomerulů anebo omezení filtrační plochy v jednotlivých glomerulech;
- Ve ztluštění nebo přestavbě glomerulárních membrán a snížení jejich permeability.
Následky každé z obou příčin jsou navzájem odlišné.
Při snížení Kf z důvodu úbytku ledvinové masy mohou zbylé nefrony, jejichž autoregulační okruhy zůstaly zachované, řídit tlakovou natriurézu stejně dobře jako před inzultem. Proto úplná ztráta části ledvinové masy (chirurgicky, traumaticky, stárnutím anebo z jiných příčin) obvykle nevyvolá hypertenzi. Stav vyústí spíše do urémie než do hypertenze. Objem ledvinového parenchymu může být redukován operací, úrazem, nádorovou destrukcí apod. Jindy může dojít ke snížení počtu glomerulů jejich zánětlivou přestavbou, aktivací růstových faktorů, patologickou proliferací mesangiálních elementů, obliterací glomerulů a glomerulosklerózou.
Zbylé nefrony jsou vystaveny větší objemové a průtokové zátěži a přetěžovány množstvím tekutiny. V typických nekomplikovaných případech je perfúze zbxylých nefronů více než dostatečná, takže hladiny reninu a angiotenzinu II zůstávají minimální. Každá z funkčních jednotek ale navíc musí převzít část exkrečních funkcí za zaniklé nefrony. Filtrační rychlost pak je v glomerulech vysoká a tubuly musí zpracovat velkou nálož Na+ a tekutin. Glomeruly i tubuly se rozšiřují, zbytňují a jejich kapacita roste. Přísun iontů k macula densa je zvýšený, v zatížených nefronech je potlačena sekrece reninu. Zpětnovazebně se zužuje aferentní arteriola. Funkční rezervy nefronů a jejich adaptace na příjem soli dosahují svého limitu. To je činí velice vnímavými na přívod NaCl. Rozvíjí se typická volumová hypertenze citlivá k příjmu soli. Při velkém denním příjmu soli nemohou zajistit vyrovnanou bilanci jinak než cestou dalšího vzrůstu glomerulární filtrace, tzn. zvýšením glomerulárního hydrostatického tlaku. Další nárůst vylučování solné nálože pak je možný jen za cenu zvýšení systémového arteriálního tlaku. Objemová, průtoková a nakonec i tlaková zátěž může vést k progresi poškození glomerulů, mikroalbuninurii a dalším změnám.
Po narození se už žádné nové nefrony netvoří a jejich počet se fyziologicky snižuje zejména po 40. roce věku. Vyhodnocení provedená v některých studiích naznačují, že hypertenze s nízkou pravděpodobností postihuje jedince s počtem glomerulů nad jeden milion v jedné ledvině. Naproti tomu jedinci s počtem glomerulů menším než 600 000 v jedné ledvině mají statisticky zřetelný sklon k hypertenzi, která je v iniciálních fázích provázena abnormálně vysokou glomerulární filtrací. Neví se, zda nízký počet glomerulů v ledvině je primárně geneticky determinován, anebo zda se dědí předpoklady k rychlejšímu úbytku glomerulů.
Po snížení Kf z důvodu poruchy permeability klesá rychlost glomerulární filtrace a filtrační frakce. Macula densa registruje nedostatečný obsah iontů v distálním tubulu a aktivuje tubuloglomerulární zpětnou vazbu. Rozšiřuje se vas afferens. Na rozdíl od situace provázející úbytek ledvinové masy roste sekrece reninu a tvorba angiotenzinu II. Tím tato hypertenze získává složku vazokonstrikční. Filtrované množství tekutiny a objem tekutiny v tubulech nakonec mohou dosáhnou hodnot potřebných pro vyrovnanou bilanci iontů a tekutin. Cenou je zvýšení sytémového arteriálního tlaku a přírůstek transkapilárního glomerulárního tlaku. Může tomu napomáhat i konstrikce eferentní arterioly. To se děje na úkor ledvinových rezerv pro vylučování NaCl. Kompenzačně zvýšený filtrační tlak v glomerulech podporuje rozvoj glomerulosklerózy, která dále sníží Kf . Může dokonce dojít k posunu funkční křivky doprava. Bludný kruh může pokračovat až k ledvinovému selhání.
Všechny hypertenze citlivé k příjmu soli se podle definice řadí k hypertenzím volumového typu. Z uvedeného rozboru vyplývá, že všechny volumové hypertenze jsou spojeny s glomerulární hypertenzí a většina z nich i s hyperfiltrací. Trvalá glomerulární hypertenze je významným rizikovým faktorem progredujícího poškození glomerulů spějícího k chronickému renálnímu selhání (obr. 10).
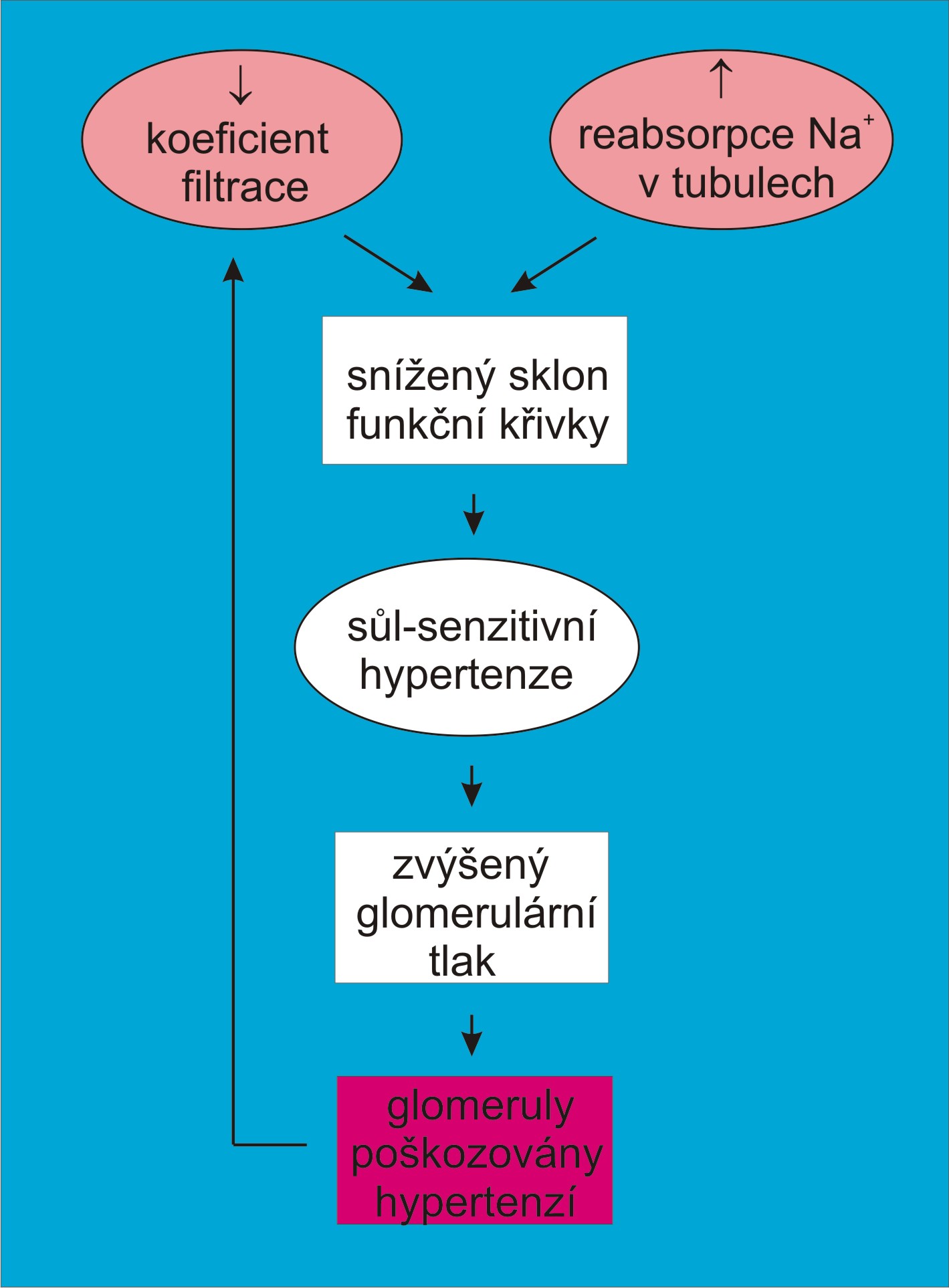
Obr. 10. Bludný kruh poškozování glomerulů při objemové hypertenzi spojené s glomerulární hypertenzí.
Vazokonstrikční hypertenze s posunutou strmou renální funkční křivkou, necitlivá k příjmu soli
Při zvýšení pregromerulárního odporu vždy dochází k posunu renální funkční křivky doprava. Bod MAPmin (obr. 8) se posunuje k vyšším hodnotám středního systémového arteriálního tlaku. Poloha MAPmin je dána rozdílem MAP a hodnotou efektivního transglomerulárního filtračního tlaku. Ovlivňují ji dvě determinanty:
- Jakýkoliv preglomerulární odpor, s nímž se krev setkává na své cestě od srdce ke glomerulům; je namístě si všimnout, že krajním případem je stenóza aortální chlopně – v tomto případě se „hypertenze“ koncentruje v levém srdci, patogenetické mechanisy však jsou stejné.
- Tlak tekutiny (primární moče) obsažené v Bowmanově pouzdře plus onkotický tlak v kapilárách glomerulů.
Abnormální rezistence může být situována kdekoliv na aortě nad odstupem renálních arterií, na renální arterii, na její interlobární segmentové větvi, interlobulárních nebo intralobulárních tepénkách anebo aferentních arteriolách. V takových případech se adekvátního perfúzního a efektivního filtračního tlaku v glomerulech dosahuje tím, že roste střední arteriální tlak v systémové cirkulaci před místem rezistence a zvyšuje se o hodnotu, kterou je zapotřebí k překonání překážky. Systémový arteriální tlak před překážkou je obětován. To přináší rizika pro srdečně cévní soustavu, avšak za stenózou jsou cévy a nefrony před zhoubným působením zvýšeného tlaku chráněny. Ve výsledku je dosažený perfúzní tlak v ledvinových cévách za překážkou právě takový, jaký je potřebný pro fyziologické funkce nefronů. Takovou hypertenzi charakterizuje křivka posunutá doprava, ale se zachovaným strmým průběhem. Poruchu od počátku charakterizuje výrazná periferní vazokonstrikce. Proto byla pojmenována vazokonstrikční hypertenze.
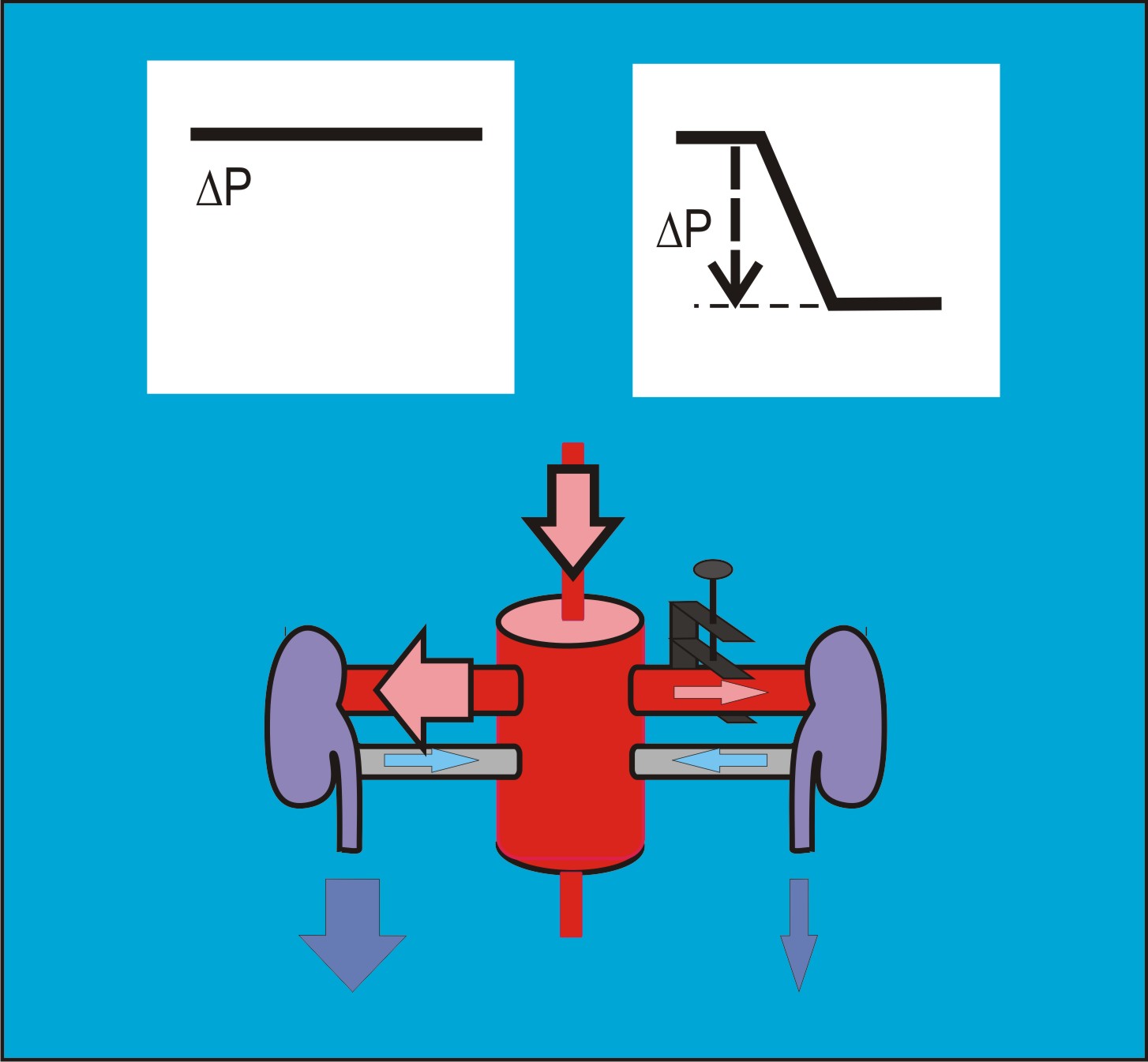
Goldblattova hypertenze 1K1C.
Hypertenzi se strmou posunutou funkční ledvinovou křivkou nejlépe objasňuje experimentální hypertenzní model typu „jedna ledvina – jedna stenóza“, 1K1C („one kidney – one clip“), který zavedl H. Goldblatt. Při tomto pokusném uspořádání se zvířeti odstranila jedna ledvina a na přívodní arterii zbývající ledviny se naložila svorka (obr. 11).
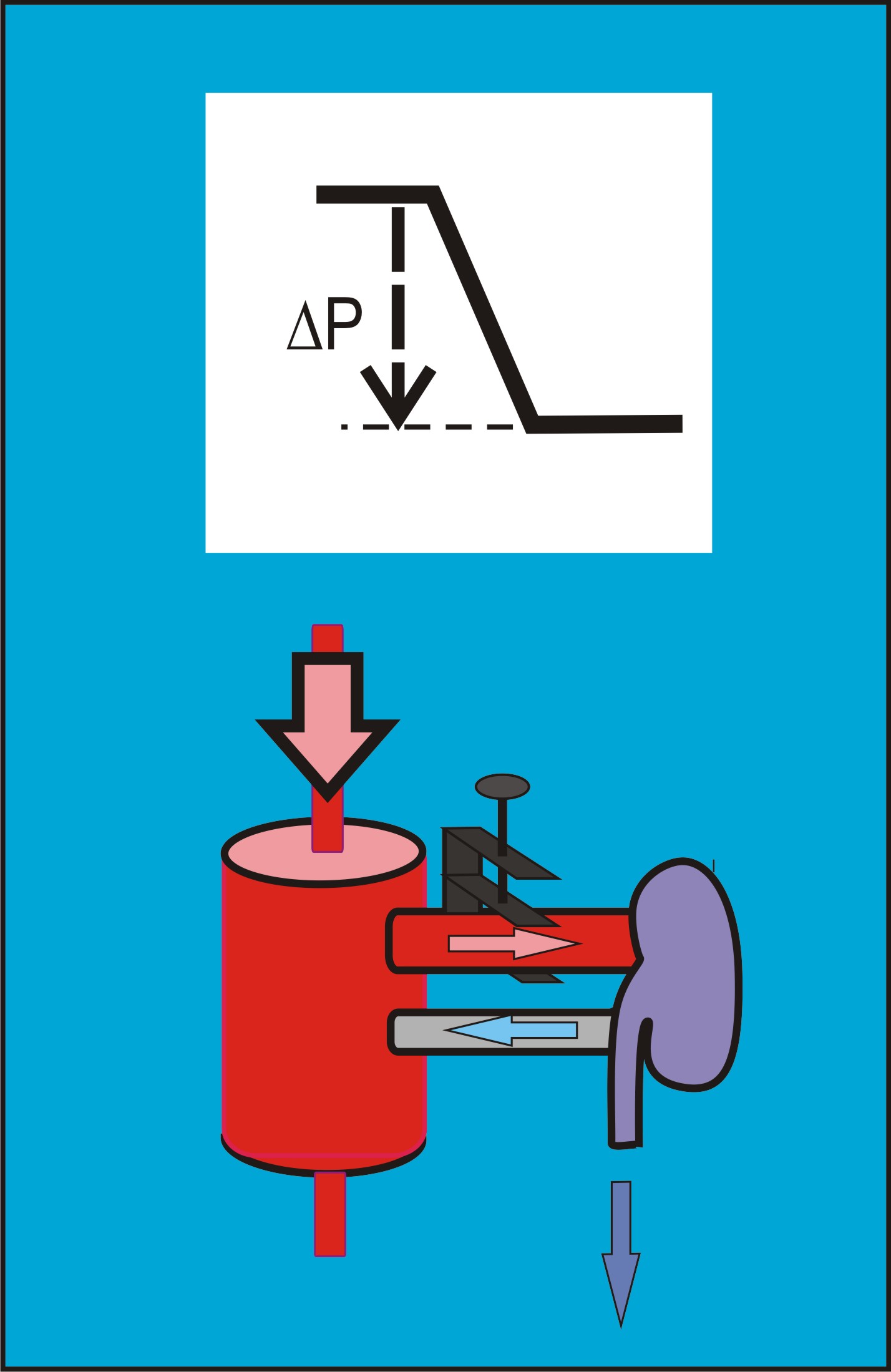
Obr. 11. Snížení hydrostatického tlaku na odporu. Model 1K1C.
Model 1K1C není samoúčelný. Reprezentuje následující klinické stavy:
- Stenózu, příp. fibromuskulární dysplázii renální arterie solitární ledviny;
- Koarktaci břišní aorty;
- Koarktaci hrudní aorty;
- (Extrapolací dojdeme ke stenóze aortální chlopně, kdy se tlak nezvyšuje v arteriálním stromu, ale v levé srdeční komoře.)
- Bilaterální pokročilou stenózu obou hlavních přívodních renálních arterií ateromy – jde o vůbec nejčastější typ renovaskulární hypertenze;
- Nadprodukci reninu z autonomně se chovajícího adenomu;
- Generalizované difuzní postižení drobných přívodních větévek (stenózami, emboliemi) v ledvinovém parenchymu obou ledvin (generalizovaný model „tisíců mikrosvorek“).
Dynamika změn. Při vývoji Goldblattovy vazokonstrikční hypertenze tyou 1K1C je vhodné rozlišovat tři stadia (obr. 12):
- Fázi prvotní (časné) vazokonstrikce;
- Vysokoreninovou natriumretenční fázi;
- Nízkoreninovou ustálenou fázi.
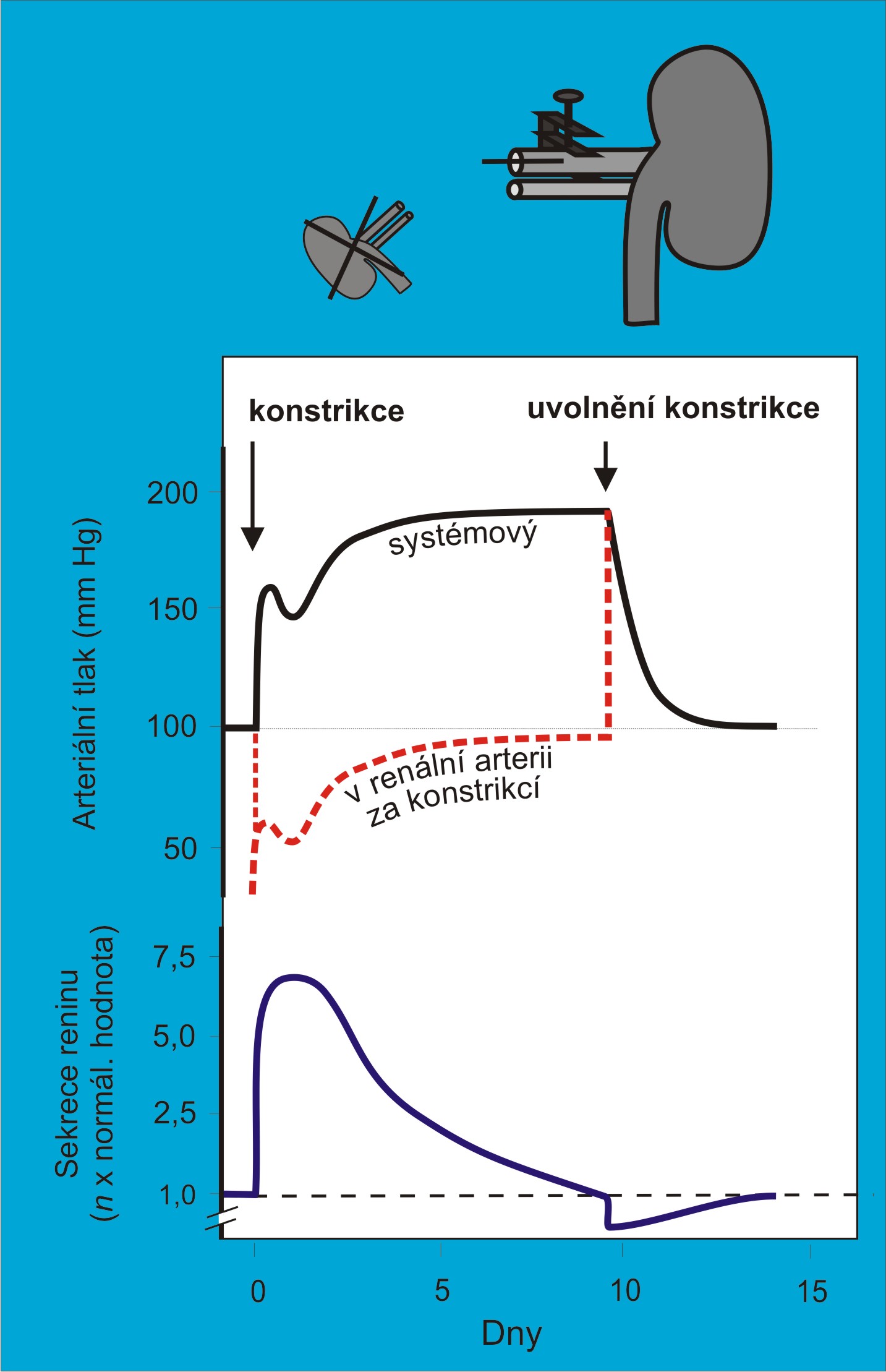
Obr. 12. Dynamika změn při vývoji Goldblattovy hypertenze typu 1K1C. (Podle AC Guyton a JE Hall: Textbook of Medical Physiology, 11th Ed. Elsevier/Saunders, Philadeplhia. 2006.)
Fáze časné vazokonstrikce. Po okluzi přívodní renální arterie se do cirkulace mohutně uvolňují vazokonstriktory. Havně jde o angiotenzin II. Jeho tvorbu spouští renin, jehož sekrece je vyprovokovaná poklesem perfúze ledvin. K vyplavení reninu dochází už v prvních desítkách minut po snížení přívodu krve do ledviny a dosahuje vrcholu za několik hodin po vzniku stenózy. Tvoří se angiotenzin II, který vyvolá rozsáhlou konstrikci jak v arteriální, tak ve venózní části cirkulace. Kapacita řečiště se tím zmenšuje, zároveň dochází k redistribuci a centralizaci oběhu, takže tlak strmě vzroste. Arteriální tlak má tendenci dosáhnout úrovně potřebné pro dostatečnou perfúzi ledviny za stenózou. Funkční křivka ledviny se prudce posunuje doprava. Zvýšení systémového arteriálního tlaku v této fázi je plně způsobeno akutním vazokonstrikčním účinkem angiotenzinu II na cévní systém. (Podobně tomu bude v případě nadprodukce reninu z autonomně se chovajícího tumoru.)
V následující vysokoreninové antriumretenční fázi, která trvá asi týden, se angiotenzin II postupně stále silněji uplatňuje přímo v ledvinovém parenchymu. Angiotenzin II zde má jak přímé sůl- a vodu-šetřící účinky, tak nepřímé sůl šetřící účinky zprostředkované aldosteronemem a sympatickým nervstvem. Renální funkční křivka se pod těmito vlivy navíc přechodně naklání doprava, k vyšším hodnotám arteriálního tlaku. To vše je provázeno retencí sodných iontů a tekutin a vzestupem středního systémového a cirkulačního plnicího tlaku.
Ustálená fáze. Na konci této druhé fáze (asi po 5 – 7 dnech) tak už je zvýšení systémového arteriálního tlaku zabezpečeno zvětšeným cirkulujícím objemem tekutin. Tlak v systémovém oběhu vzrostl do úrovně, která zajistí potřebný perfúzní tlak v ledvině. Poté už zvýšené hladiny reninu a angiotenzinu II nejsou zapotřebí, a vracejí se k normě. Renální funkční křivka znovu získává svou strmost, ale zůstává posunuta doprava (obr. 13).
Z popisu dynamiky je zřejmé, že pro vznik ustálené vazokonstrikční hypertenze 1K1C je rozhodující vystupňované natrium-retenční působení angiotenzinmu II přímo v ledvinovém parenchymu. Právě toto působení způsobí akumulaci tekutin a vede k chronickému zvýšení systémového arteriálního tlaku. Sama periferní vazokostrikce, k níž dochází v počáteční fázi vývoje hypertenze 1K1C, je jen pohotovostní, akutní reakcí, která by izolovaně, bez natriumretenčního působení angiotenzinu II v ledvinách, chronickou hypertenzi nezpůsobila (obr. 2).
Obr. 13. Posun ledvinové křivky doprava v ustálené nízkoreninové Goldblattově hypertenzi typu 1K1C. (Podle AC Guyton a JE Hall: Textbook of Medical Physiology, 11th Ed. Elsevier/Saunders, Philadeplhia. 2006.) Schéma je možno zhlédnout i v animované podobě.
Shrnutí vývoje hypertenze 1K1C. Hypertenze v systémovém oběhu modelu 1K1C dosáhne právě takové hladiny, při níž krevní tlak a průtok krve za stenózou (svorkou) zaručí vyrovnanou bilanci solí a tekutin. Renální funkční křivka ledviny za stenózou je posunuta k vyšším hodnotám systémového arteriáního tlaku (měřeného na paži). Je významné, že pokud zůstávají funkční rezervy ledvinového parenchymu neporušeny, křivka si zachovává svou strmost (obr. 13). Regulace kolem nově ustálené hodnoty tlaku pak je stejně výkonná jako regulace při normálním tlaku. Změnila se jen regulovaná hladina.
Hodnoty krevního tlaku, reprezentované ve variantě 1K1C strmou posunutou křivkou, jsou po ustálení hypertenze necitlivé k příjmu soli. Navíc stojí za zmínku, že objem cirkulující tekutiny a srdeční výdej nemocných se prakticky neliší od noremálních hodnot.
Hypertenze 1K1C snad ze všech typů hypertenze nejnázorněji ilustruje, jak se arteriální krevní tlak v systémové části cévního řečiště před stenózou obětuje požadavku zajistit vyrovnanou objemovou bilanci tekutin. Ischemizovaná ledvina dokáže vnutit celé cirkulaci novou úroveň tlaku. Disponuje tak výkonnými autoregulačními mechanismy, že cirkulaci přiměje zvýšit tlak před stenózou na úroveň, kdy poměry za stenózou budou přesně odpovídat jejím funkčním požadavkům. Celá cirkulace trpí vysokým tlakem, ledvina za stenózou - na rozdíl od situace při volumové hypertenzi – však je před hypertenzí chráněna (obr. 11, obr. 14).
Obr. 14. Systémovou Goldblattovu hypertenzi neprovází glomerulární hypertenze. Glomeruly za stenózou jsou před hypertenzí chráněny.
Klinické souvislosti. Dokud se postižení omezuje jen na přívodní krevní cesty a funkční rezervy samotného ledvinového parenchymu zůstávají intaktní, ledvina je po dosažení dostatečného perfúzního tlaku za stenózou schopna regulovat tlak v cirkulaci stejně účinně, jako kdyby šlo o normální tlakovou hladinu (obr. 13). Z obrázku 12 je patrné, hypertenze 1K1C je ve své ustálené formě nízkoreninovou hypertenzí s nízkými hladinami angiotenzinu II. Pacienti tedy nemohou těžit z prostého omezení příjmu soli. Strmá ledvinová křivka tuto hypertenzi charakterizuje jako necitlivou k soli. Pacienti jsou schopni vyloučit i velká kvanta soli bez dalšího významného vzestupu arteriálního tlaku. Naproti tomu při depleci soli anebo krevního objemu (snížením příjmu anebo zvýšením ztrát, podpořenými např. diuretiky), kdy tlak může poklesnout, se zdánlivě paradoxně (při vysokém tlaku!) objeví známky aktivace renin-angiotenzinového systému. Léčba této hypertenze proto musí zahrnovat inhibici renin-angiotenzinového systému.
Podobně jako u volumové hypertenze zde platí, že objem krve při vazokonstrikční hypertenzi není úměrný kapacitě krevního řečiště. K tomu by mohlo dojít jen v případě, že by se veškeré účinky reninu a angiotenzinu II v dynamické fázi vývoje této hypertenze vyčerpaly pouze periferním vazokonstrikčním působením mimo ledviny, bez ovlivnění exkrece Na+. Angiotenzin však má kromě svých periferních vazokonstrikčních účinků robustní vliv na zpětné vstřebávání iontů Na+ v ledvinách (obr. 15). Patří k nejvýkonnějším stimulátorům reabsorpce sodných iontů v proximálních tubulech. Navíc podněcuje sekreci aldosteronu v nadledvinách, který zasahuje v distálních tubulech.
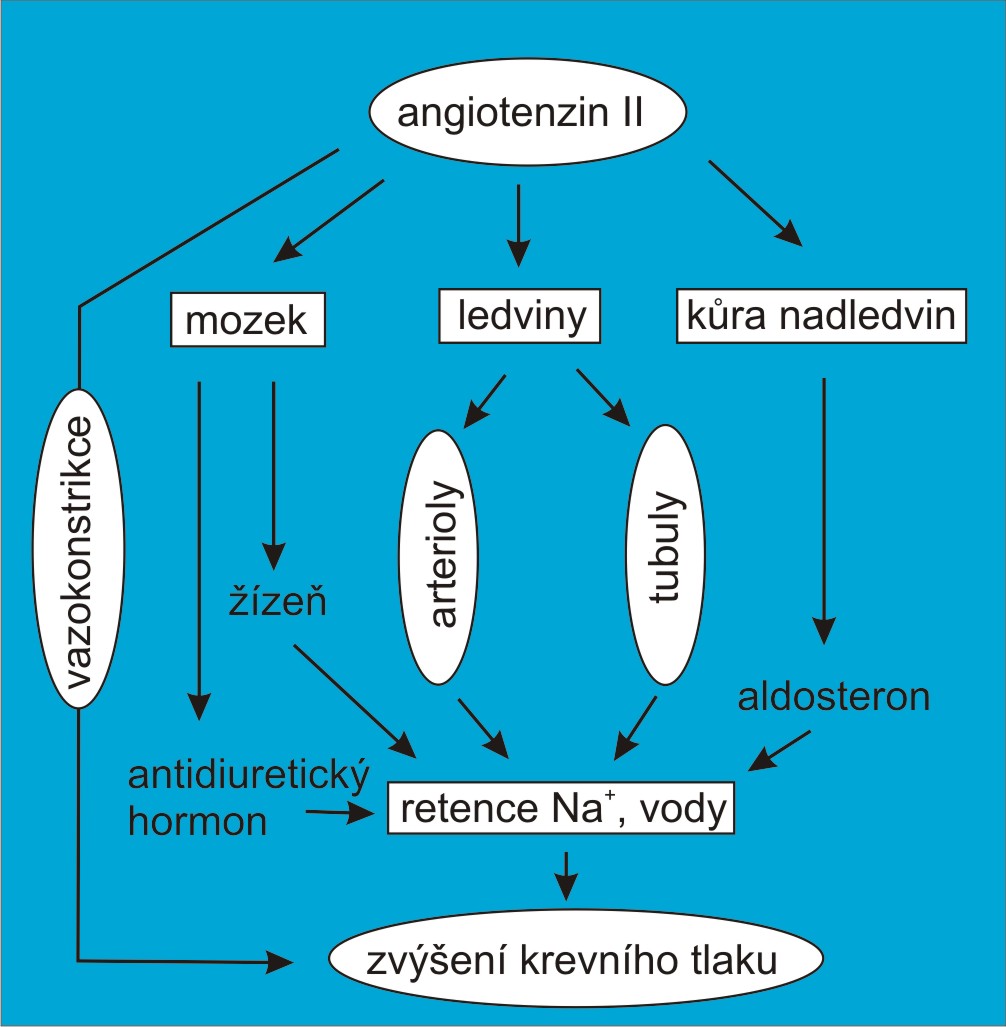
Obr. 15. Schéma působení angiotenzinu na regulaci krevního tlaku. Působení angiotenzinu zahrnuje jednak krátkodobé, pohotovostní efekty (arteriolokonstrikce a venokonstrikce), jednak dlouhodobé vlivy na ledvinovou funkční křivku. Angiotenzin je nejmocnějším stimulátorem retence Na+; na rozdíl od aldostreronu se hlavně uplatňuje v proximálních tubulech. (Podle J Veselý: Tlaková diuréza a arteriální hypertenze. Epava, Olomouc, 2002.)
Jakkoli to tedy může znít paradoxně, je rozvinuté stadium vazokonstrikční hypertenze hypertenzí z nadbytku tekutiny. Je obtížné dostatečně zdůraznit, že vlastním patofyziologickým podkladem trvalé hypertenze je i zde – stejně jako u volumové hypertenze – relativní zvětšení náplně vzhledem ke kapacitě cirkulace a nárůst cirkulačního plnicího tlaku. Příčinou tohoto stavu je alterace funkční křivky ledvin.
Smíšené hypertenze s kombinovaným postižením funkční renální křivky
Goldblattova hypertenze 2K1C.
Hypertenzi s kombinovaným postižením, kdy křivka je zároveň posunuta a má i alterovanou směrnici, nejlépe reprezentuje Goldblattův experimentální hypertenzní model „dvě ledviny-jedna stenóza“, 2K1C („two kidney – one clip“) (obr. 16). Jde o model se dvěma ledvinami, z nichž pouze jedna je ischemická. Tento model se zásadně liší od 1K1C v tom ohledu, že poté, co se hladina reninu činností ischemické ledviny zvýší, nikdy se nevrátí ke své předchozí normální úrovni.
Obr. 16. Ilustrace modelu 2K1C.
Klinickými variantami Goldblattova modelu 2K1C jsou následující stavy:
- Unilaterální stenóza na podkladě aterosklereózy nebo fibromuskulární dysplázie jedné hlavní přívodní renální arterie při přítomnosti obou ledvin.
- Segmentová arteriální stenóza v solitární ledvině.
- Okluze (stenóza, embolie) drobných přívodních větévek v omezeném segmentu nebo v několika segmentech ledvinového parenchymu v jedné nebo v obou ledvinách (segmentová varianta modelu „tisíců mikrosvorek“ ).
Odlišnost modelu 2K1C od modelu 1K1C. Hypoperfúze ledviny za svorkou v modelu 2K1C vyvolá aktivaci renin-angiotenzinového systému stejně jako v modelu 1K1C. Snahy ischemické ledviny zvýšit plnicí a arteriální tlak však ve značné míře neutralizuje druhá, zdravá ledvina. Ta je při zvýšeném tlaku hyperperfundována. Zdravá ledvina neustále odvádí část tekutiny akumulované ischemickou ledvinou, a pracuje tak proti jejím požadavkům. Arteriální tlak v modelu 2K1C proto nikdy nedosáhne takové výše, aby uspokojil nároky ischemizované ledviny za stenózou. Nedostatečný průtok tekutiny v nefronech za stenózou bude neustále stimulovat výdej reninu. Proto se reninová aktivita a hladina angiotenzinu II v modelu 2K1C nikdy nevrátí na výchozí normální hodnoty. V rozvinutém stadiu půjde o hypertenzi s vysokou hladinou reninu a angiotenzinu II.
Angiotenzin svými silnýnmi Na+-šetřícími účinky výrazně omezuje strmost ledvinové funkční křivky v obou ledvinách. Výsledná hypertenze bude kombinací hypertenze vazokonstrikčního a objemového typu a bude omezeně sůl-senzitivní. Její renální funkční křivka bude trvale nejen posunuta, ale i skloněna k ose arteriálního tlaku (obr. 17).
Obr. 17. Skloněná ledvinová křivka posunutá doprava v ustálené vysokoreninové Goldblattově hypertenzi typu 2K1C. (Podle AC Guyton a JE Hall: Textbook of Medical Physiology, 11th Ed. Elsevier/Saunders, Philadeplhia. 2006.)
Hypotéza „tisíců mikrosvorek“ – zvýšený odpor malých intraparenchymálních aferentních cév
Aterosklerotické a trombotické změny mohou postihovat nejen velké, ale i segmentové anebo drobné arterky v ledvinách. V ledvinách pacientů s mírnou nekomplikovanou hypertenzí se podařilo vedle sebe prokázat políčka s aferentními arteriolami, které měly normální průběh a průřez, a políčka, kde byly aferentní arterioly zřetelně zúženy koncentrickou hypertrofií stěn. Nefrony situované za takto postiženými arteriolami nemohou být zásobeny dostatečným přívodem krve. Ischemická políčka jsou stimulována k produkci reninu a tvorbě angiotenzinu II. Patofyziologické chování takové ledviny odpovídá klasickému modelu „dvě ledviny – jedna stenóza“. Představu jedné svorky na velké tepně zde nahrazuje představa „tisíců mikrosvorek“ rozmístěných na malých arteriích v ohraničených segmentech ledvinového parenchymu. Podobné postižení ledvinového parenchymu vede k hypertenzi smíšeného typu se střední až vysokou plazmatickou reninovou aktivitou.
Poruchy aferentních arteriol nemusí být vždy způsobeny jen strukturními, organickými příčinami. Může jít i o funkční změny. U části jedinců s predispozicí k hypertenzi se prokazuje nedostatečná vazodilatační odpověď v ledvinách na zvýšený příjem soli. Porucha může být způsobena nadměrnou lokální aktivitou vazokonstrikčních faktorů v ledvinách (např. angiotenzinu II) (obr. 3, vlevo), nedostatečnou tvorbou vazodilateačních faktorů (např. dopaminu) (obr. 3, vpravo), nebo může jít o patologicky zvýšenou vnímavost cév na vazokonstrikční podněty, jako je tomu například při genetickém zmnožení α-adrenergních receptorů (popsáno níže). Představa „tisíců mikrosvorek“ ukazuje, jak může k závažným poruchám regulace krevního tlaku a vzniku hypertenze dojít na základě drobných, klinicky jen obtížně detekovatelných patologických alterací.
Ledviny jsou nejen původcem hypertenze, ale spolu s mozkem a srdcem také jsou jedním z jejích nejvýznamnějších cílových orgánů. Hypertenzi provázejí sekundární změny ledvinového parenchymu. Už pro hypertenze mírného stupně platí, že při nich více roste ledvinová cévní rezistence než celková systémová periferní rezistence. Mezi středním systémovým arteriálním tlakem a odporem aferentní arterie existuje výrazná pozitivní korelace. Sekundání zúžení preglomerulárních arterií způsobuje posun ledvinové funkční křivky doprava. I hypertenze, která původně začala jso sůl-senzitivní, se tak ve svém průběhu mění ve formu hypertenze s přídatnou sůl-nesenzitivní složkou. Právě smíšené hypertenze pravděpodobně představují největší počet hypertenzí v pokročilých stadiích. Sekundárními změnami preglomerulárních arteriol a glomerulů lze současně vysvětlit, proč pokročilou hypertenzi, byť začala jako sůl-senzitivní, se stěží podaří vyléčit pouhým omezením příjmu soli. Kombinované postižení ledvinové funkční křivky zřejmě zároveň představuje zásadní problém populačních studií prováděných mezi hypertoniky s cílem vyhodnotit jejich citlivost k příjmu soli. Mnohem větší pravděpodobnost záchytu citlivosti k soli má vyšetření v počátečních fázích hypertenze, před tím, než dojde k překrytí jejích základních rysů sekundárními průvodními změnami.
Neurogenní hypertenze
Přestože se úloha nervového systému v rozvoji hypertenze diskutuje po několik desítek let, údaje dosud nedovolují formulovat ucelenou teorii neurogenní hypertenze. Teorie musí respektovat skutečnost, že jediná cesta k chronické systémové arteriální hypertenzi vede přes alteraci ledvinové funkční křivky.
Nervové podněty mohou trvale ovlivnit ledvinovou křivku za předpokladu:
- Dlouhodbě zvýšené aktivity nadřazených nervových center;
- Nepřiměřené lokální citlivosti ledvinového parenchymu na normální vzruchovou aktivitu v sympatických nervech nebo na přítomnost normálních hladin transmiterů.
Dlouhodobé dráždění eferentních sympatických renálních nervů
Intenzivní sympatická aktivita mění průběh ledvinové funkční křivky. Zdroje nadměrné aktivity, odkud sympatikus může významně přispět k vývoji hypertenze, lze lokalizovat do čtyř úrovní:
- Aferentního signalizačního článku z receptorů v cirkulaci anebo z cirkumventrikulárního organum vasculosum;
- Řídících center v mozku, zejména předního jádra hypotalamu;
- Eferentního článku zajišťujícího inervaci cév a ledvin;
- Receptorů sympatiku nacházejících se v cílové tkáni.
Mechanismy lokalizované v první úrovni (aferentní článek) se zdají být hlavně krátkodobé. Mechanismy uvedené ve druhé a třetí úrovni zatím nejsou příliš jasné. Obecně se ale přijímá, že v centrálním nervovém systému se může vytvořit stálé ohnisko podráždění a zdroj zvýšené impulzace, která se pak může trvale šířit renálními sympatickými nervy. Opakované anebo trvalé dráždění center tak může způsobit nežádoucí změny funkční ledvinové křivky. V této souvislosti je namístě zmínit imidazolinové receptory v centrálním nervovém systému.
Imidazolinové receptory v centrálním nervovém systému. Neurony horní (rostrální) ventrolaterální medulární vazomotorické formace (RVLM), která má presorické funkce, jsou na svých povrchových membránách vybaveny imidazolinovými receptory I1 (obr. 18). Jejich přirozené ligandy dosud nejsou s jistotou známy. Nereagují s katecholaminy. Jejich stimulace umělými farmakologickými agonisty inhibuje sympatikus a navozuje hypotenzi. Jejich nedostatečné působení v RVLM by tak mohlo být spoluodpověné za vznik hypertenze.
Uměle syntetizovaní agonisté I1-receptorů, jako jsou moxonidin anebo rilmenidin, jsou poměrně novou nadějnou skupinou antihypertenziv. Působí i na periferní I1-receptory v ledvinách. Stimulací ledvinových I1-receptorů přímo zvyšují natriurézu a diurézu. Jiné centrální antihypertenzivum, clonidin, imidazolinový preparát, který rovněž stimuluje I1-receptory, právě uvedenou výhodu nemá, protože je zároveň poměrně účinným agonistou α2-receptorů. Zatímco centrálně tlumí sympatikus (a má i sedativní účinky), v ledvinách působí antinatriureticky a antidiureticky.
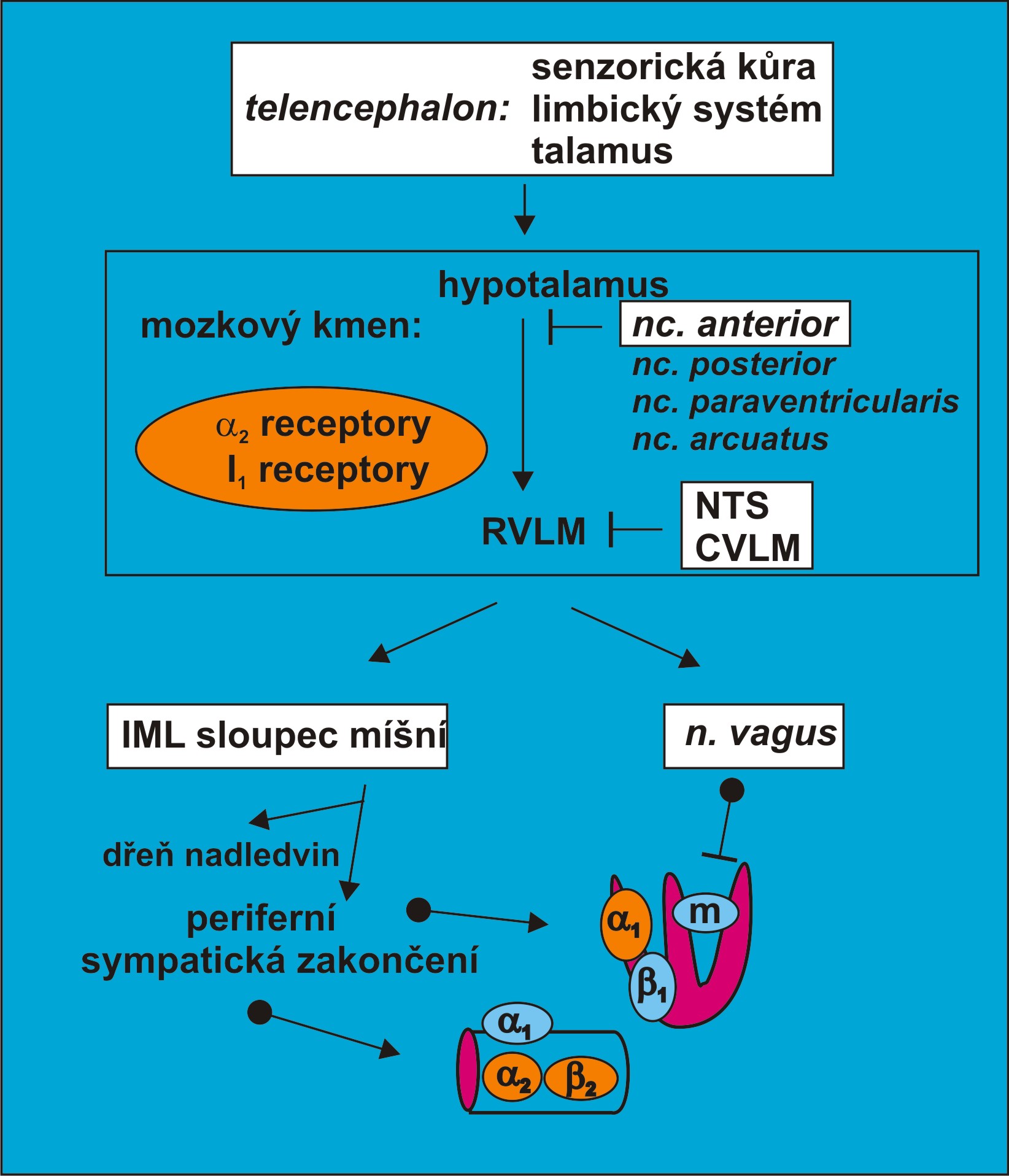
Obr. 18. Schéma regulace oběhového systému autonomním nervovým sympatickým systémem s jeho hlavními receptory. (Podle J Veselý: Tlaková diuréza a arteriální hypertenze. Epava, Olomouc, 2002.)
Genetické abnormality adrenergních receptorů v ledvinách
Geneticky determinované zmnožení α-adrenoreceptorů anebo zvýšení jejich reaktivity v ledvinách se považuje za jednu z velmi pravděpodobných příčin patologického sklonu renální funkční křivky a esenciální hypertenze. Sympatickými α1 receptory je hojně vybavena aferentní arteriola (obr. 19). Anormálně početné α-receptory zprostředkují masivní vazokonstrikci v ledvinách už při fyziologických, nezvýšených koncentracích neurotransmiterů, a způsobí alteraci renální funkční křivky. Diagnosticky je významné, že abnormality α-receptorů se neomezují jen na ledviny. Manifestují se i v jiných tkáních.
Stimulace α1receptorů farmakologickými agonisty zvyšuje u pacientů s rodinnou zátěží arteriální tlak více než u jedinců anamnesticky negativních. Tyto známky jsou přítomny i u zatím zdravých potomků hypertenzivních rodičů. Uvedené skutečnosti podporují výzkum genů α-receptorů jako kandidátních genů hypertenze. Vyšší hustota α-receptorů zatím nebyla prokázána u sekundárních hypertenzí.

Obr. 19. Adrenergní a angiotenzinová receptorová výbava přívodních a odvodných glomerulárních cév a tubulů. Bližší údaje jsou uvedeny v textu. (Podle J Veselý: Tlaková diuréza a arteriální hypertenze. Epava, Olomouc, 2002.)
Epitel proximálních ledvinových kanálků je vybaven receptory α2 (obr. 19). Sympatikus svými účinky přímo stimuluje reabsorpci Na+. Pro diagnostické účely je důležité, že část pacientů s primární hypertenzí také má zmnoženy α2-receptory v trombocytech. Agonisté centrálních α2-receptorů, jako např. clonidin nebo guanfacin, slouží jako hypotenziva; clonidin nicméně svým periferním působením stimuluje i reabsorpci Na+. Pokud jde o funkce v nervovém přenosu, receptory α2 jsou uloženy na presynaptických zakončeních. Jejich inhibice napomáhá exocytóze a zpomaluje zpětné vychytávání mediátorů. V souladu s tím podání farmakologických antagonistů α2-receptorů (yohimbin) výrazně zvýší krevní tlak přinejmenším u části hypertoniků. Tento účinek se nejspíše opírá o působení antagonistů v centrálním nervovém systému.
Některé primární změny se podařilo prokázat také v případě β2-adrenergních receptorů. Obecně se však změny hustoty β-receptorů v srdci, v cévách a v ledvinách považují spíše za sekundární následky hypertenze, než aby hráli primární příčinnou roli. Při dlouhodobé hyperaktivitě sympatiku dochází k desenzitizaci a snížení hustoty jak α-, tak β-receptorů. To značně komplikuje laboratorní i klinický výzkum, zejména pokud jde o pokročilé fáze hypertenze.
Genetické abnormality metabolismu dopaminu a dopaminergních receptorů v ledvinách
Dopamin je fylogeneticky starý mediátor a odpovídá asi za 80 % vylučovací kapacity ledvin pro Na+. K hypertenzi mohou vést tři hlavní defekty defekty dopaminergního systému v ledvinách:
- Deficit produkce dopaminu;
- Porucha působení dopaminu na cévy;
- Porucha působení dopaminu v tubulech.
Produkce dopaminu v ledvinách klesá s věkem. Navíc může trpět při různých chorobách (např. diabetu). Deficit produkce dopaminu může být způsoben sníženou aktivitou transportního systému obstarávajícího přísun DOPA do proximálních tubulárních buněk anebo sníženou aktivitou DOPA-dekarboxylázy v buňkách. U postižených jedinců potom nedochází k očekávanému fyziologickému zvýšení produkce dopaminu po zátěži zvýšeným příjmem soli. Také hustota dopaminergních receptorů D1, které v ledvinových tubulech zprostředkují natriurézu a diurézu, a tvorba cAMP po dopaminomimetických podnětech v ledvinách se pronikavě snižují s věkem. Ve stáří se tedy postupně vytrácí normální příznivé působení dopaminu na renální funkční křivku. Dochází k omezení natriuretické kapacity. Nepřiměřený přívod soli potom může napomáhat poruchám bilance iontů a tekutin a poruchám regulace úrovně arteriálního tlaku.
Poměrně dobře prozkoumán je defekt postreceptorové signalizace membránových D1-receptorů tubulárních buněk. Spočívá v odpřažení receptorů od G-proteinů. Velké množství nadměrně fosforylovaných D1-receptorů pak zůstává v cytoplazmě a není k dispozici pro interakce na membránách. Následkem je omezení natriurézy.
Hyperinzulinémie a diabetes
Hyperinzulinémie neovlivňuje renální funkční křivku. Údaje o úloze inzulínu ve vývoji hypertenze jsouo rozporné. Kauzální působení inzulínu, případně hyperinzulinémie, při vzniku hypertenze u lidí není doložena. Pacienti s inzulinomy netrpí vždy hypertenzí. Inzulín nepodporuje hypertenzivní vlivy angiotenzinu II nebo noradrenalinu. Naopak, je to angiotenzin II, jeden z ústředních patogenetických faktorů hypertenze, jenž může navodit inzulinovou rezistenci. Inzulín neovlivnil tlak nebo sympatickou aktivitu, když byl po několik dní podáván infuzí přímo do mozkové cirkulace. Není žádný důkaz, že by hyperinulinémie měla přímý vliv na renální funkční křivku. Hyperinzulinémie experimentálně udržovaná infúzí po několik týdnů nezvyšuje arteriální tlak ani u psa, ani u člověka. Spíše vyvolává vazodilataci a tlak poněkud snižuje.
Obezitu, která je hlavním rizikovým faktorem hypertenze, téměř vždy provází hyperinzulinémie. Existující korelace mezi hladinou inzulínu v plazmě a krevním tlakem tak může být přítomna prostě proto, že obezita sice zvyšuje krevní tlak a navozuje inzulinorezistenci, ale oba tyto důsledky obezity jsou paralelní a jejich mechanismy jsou z velké části vzájemně nezávislé. U obézních osob se častěji vyskytuje nejen hypertenze, ale i glukózová intolerance a diabetes. Mezi osobami dlouhodobě nemocnými cukrovkou se hypertenze vyskytuje ve 40 – 80 %. U diabetiků typu 1 koreluje výskyt hypertenze s délkou onemocnění, u diabetiků typu 2 ne. Redukce tělesné hmotnosti často vede nejen k úpravě diabetu, ale i hypertenze. Diabetes je jedním z hlavních rizikových faktorů aterosklerózy a může podporovat vznik aterosklerotických lézí v přívodních cévách a v ledvinové mikrocirkulaci, nebo poškození glomerulů. Diabetes je dnes nejčastější příčinou chronického selhání ledvin, hypertenze ho ve statistice selhání ledvin následuje. Glukóza a produkty neenzymové glykace jsou výkonnými lapači radikálu oxidu dusnatého NO. Glukóza tak může způsobit endotelovou dysfunkci anebo se může se podílet na jejím dalším zhoršování.
Existuje představa, podle níž se na expanzi extracelulárního prostoru a vzniku hypertenze u diabetu může podílet hyperglykémiea z ní plynoucí vysoká nálož glukózy v glomerulárním filtrátu. Glukóza se v proximálních tubulech reabsorbuje elektrogenním Na+-dependentním transportním systémem. Množství natriových iontů, provázejících její pohyb, by mohlo stimulovat Na+/K+-ATPázu bazolaterálních membrán ke zvýšenému transportu Na+ do intersticia. Po dosažení tubulárního transportního maxima a následné osmotické diuréze však bývá hyperglykémie spojena se zvýšenými ztrátami vody. Existují tak podmínky pro sekundární aldosteronismus a vývoj hypovolemické hypernatrémie. Hypernatrémie má tonizující vliv na sympatikus. Nakolik by se tyto faktoy mohly kauzálně uplatnit při vývoji hypertenze, není s jistotou známo.
Klinická charakteristika rozvinuté (pokročilé) hypertenze
Rozvinutá systémové arteriální hypertenze zahrnuje následující charakteristiky (obr. 20):
- Chronicky zvýšený systémový arteriální tlak;
- Zvýšený celkový periferní odpor v systémovém oběhu;
- Normální anebo téměř normální srdeční výdej;
- Přestavbu stěn cév;
- Přestavbu mikrocirkulace;
- Poškození parenchymu cílových orgánů, zejména mozku, ledvin a srdce.

Obr. 20. Srdeční výdej a periferní rezistence v jednotlivých stádiích vývoje hypertenze. (Podle Ventura O, Taler SJ, Strobeck JE: Am. J. Hypertens. 18, 26S-43S, 2005.) Pro porozumění je užitečné srovnat tento obrázek s obr. 4 a obr. 12.
Změny cév
V cévách jsou výrazné alterace jak na úrovni velkých cév (makroangiopatie), tak na úrovni drobných cév a mikrocirkulace (mikroangiopatie).
- Makroangiopatie mají následující hlavní formy:
- Sonograficky zjistitelné ztluštění arteriální stěny;
- Tloušťka intimy-medie karotid ≥ 0,9 mm;
- Tvorba aterosklerotických plátů – hypertenze je významným rizikovým faktorem aterosklerózy.
- Mikroangiopatie přináší následující hlavní patologické změny:
- Nefropatie;
- Glomeruloskleróza, selhání ledvin. Pokročilá systémová arteriální hypertenze je druhou hlavní příčinou chronického selhání ledvin (po diabetu).
- Oční postižení: Retinopatie, hemoragie, exsudáty, edém papily při pokročilém postižení.
- Redukce hustoty malých cév a mikrocirkulace v periférii.
- K této přestavbě dochází sekundárně, následkem zvýšeného arteriálnícho tlaku a zvýšeného krevního průtoku tkáněmi. Ve svých důsledcích je to právě přestavba mikrocirkuklace, která je odpovědná za přítomnost chronicky zvýšeného celkového periferního odporu při arteriální hypertenzi. Na rozdíl od dřívějších dlouho tradovaných představ je dnes zřejmé, že chronicky zvýšený celkový periferní odpor provázející arteriální hypertenzi je důsledkem hypertenze, nikoliv její primární příčinou.
Změny ledvin
Podle dnešních znalostí jsou ledviny primárním orgánem odpovědným za hypertenzi. Na druhé straně jsou samy hypertenzí poškozovány. Hypertenze spolu s diabetes mellitus představují dvě nejčastější příčiny chronického selhání ledvin. Za progresi změn je především odpovědná mikroangiopatie. Patologicko-anatomicky má formu glomerulární nefropatie (nefrosklerózy). S poškozením glomerulů souvisí proteinurie. Nejprve má formu mikroalbuminurie (30 – 300 mg/24 hod); později přechází v manifestní proteinurii (> 300 mg/24 hod). Rutinním klinickým ukazatelem progredujícího omezení ledvinových fukcí je vzestup koncentrace plazmatického kreatininu (M > 130 μmol/l, Ž > 124 μmol/l).
Změny mozku
Změny mozkových cév se nejčastěji se manifestují cévní mozkovou příhodou. Cévní mozkové příhody jsou nejčastěji ischemické (asi z 85 %). Často mohou mít formu přechodných ischemických atak (TIA). Hemoragické příhody tvoří asi 15 %. Za součást změn nervové tkáně je možno považovaat i výše uvedenou retinopatii anebo krvácení do sítnice.
Změny srdce
Srdce se při hypertenzi pronikavě mění. Rozvíjí se hypertrofie levé komory (EKG: > 38 mV; echokardiogram: index hmotnosti komory – M ≥ 125 g/m2, Ž ≥ 110 g/m2). Hypertenze a hypertrofie jsou významné rizikové faktory pro vznik ischemické nemoci srdeční a infarktu myokardu. Konečným stadiem nemoci může být srdeční selhání.
Hypertenzní krize
Hypertenzní krize je urgentním stavem s náhlým a výrazným vzestupem diastolického arteriálního tlaku, často nad 130 – 140 mm Hg. Může vzniknout v kterékoliv fázi hypertenze, nemusí jít jen o hypertenzi už provázenou pokročilými změnami. Pacienta ohrožuje rychle progredující poškození cílových orgánů (jednoho nebo několika):
- Hypertenzní encefalopatie;
- Retinopatie až neuroretinopatie;
- Selhání levé komory;
- Možnost aortální disekce;
- Renální selhání.
Hypertenzní encefalopatie
Postihuje asi 1 % nemocných. Má následující znaky:
- Prudké zvýšení systolického a diastolického tlaku;
- Tlak zvyšuje průtok krve mozkem;
- V mozkovém řečišti selhávají prekapilární odpory;
- Dochází k poškození cévní stěny, poškození kapilár, mění se propustnost kapilár;
- Tekutina uniká z cév do intersticia;
- Vzniká mozkový edém a intrakraniální hypertenze.
- Rozvíjí se syndrom hypertenzní encefalopatie. Neurologické symptomy encefalopatie se objevují náhle, a pokud se tlak razantně nesníží, dramaticky progredují.
- Hrozí krvácení do mozku.
Syndrom hypertenzní encefalopatie má následující klinické příznaky:
- Bolesti hlavy, zmatenost, somnolence, stupor (postiženého možno probudit jen velmi silnými bolestivými podněty);
- Poškození až ztráta vidění;
- Přechodné příznaky ložiskového poškození mozku (přechodná amaurosa – slepota)
- Retinopatie – krvácení do sítnice a extravazáty.
Úspěšnost současné léčby hypertenze
Léčba hypertenze představuje nejen obrovský zdravotnický, ale také ekonomický problém. Americké zdroje z roku 2004 uvádějí, že každý zaměstnaný Američan přispívá na léčebné výlohy spojené s léčbou hypertenze částkou 400 dolarů za rok. Přesto se snížení tlaku do žádoucích mezí dosahuje pouze asi u 25 % léčených. To je nedostatečné jak z hledisek zdravotnických, tak ekonomických. Nelze to vysvětlovat jen špatnou spoluprací (compliance) nemocných. Přes veškerý dosavadní pokrok je potřeba mít na paměti, že stále ještě neznáme terapii, která by byla dostatečně účinná u všech, kdo ji potřebují. Usilovně se hledají další cesty zlepšení terapeutických přístupů (obr. 21). Populační studie ukazují, že snížení systolického tlaku o 10 mm Hg sníží mortalitu na mozkovou mrtvici o 40 %. Už snížení systolického tlaku o 2 mm Hg vede ke snížení mortality na mozkovou mrtvici o 10 %.
Obr. 21. Přínos impedanční kardiografie ke stanovení strategie při léčbě hypertenze. (Podle Ventura O, Taler SJ, Strobeck JE: Am. J. Hypertens. 18, 26S-43S, 2005.)
Historie
- 1856 – L. Traube
- 1900 – E.H. Starling, F. Volhard, T. Fahr, W. M. Bayliss, H. Goldblatt. Zdůrazňovali primární úlohu ledvin při vzniku hypertenze.
- 1843 – 1854: C. Ludwig, F. Goll. Odhalili a popsali jev tlakové diurézy.
- 1950 – J. M. Ledingham, R. D. Cohen, J. G. G. Borst, A. C. Guyton. Dále významně rozpracovali koncepci opírající se o primární etiopatogenetické postavení ledvin při vzniku hypertenze.
- 1872 – 1879: W. W. Gull, H. G. Suton, F. A. Mahomed a po nich zejména
- 1936 – Sir G. W. Pickering – prosazovali mylnou koncepci zakládající se na primární úloze změn periferního odporu při vzniku systémové arteriální hypertenze.
Při tvorbě této stránky byl se svolením autora a vydavatele částečně využit text a obrazová dokumentace monografie „Tlaková diuréza a arteriální hypertenze“ (Autor: J. Veselý. Vydavatelství EPAVA, Olomouc. 2002). V případě zájmu o původní širší text prosím kontaktujte vydatelství EPAVA, Olomouc (E-mail: ec@epava.cz).
Zpracovali: Jaroslav Veselý, Ústav patologické fyziologie LF UP; a Jan Václavík, I. interní klinika LF UP a FN v Olomouci
Literatura k dalšímu studiu:
Cífková R, Škodová Z, Bruthans J, Holub J, Adámková V, Jozífová M, Galovcová M, Wohlfahrt P, Krajčoviechová A, Petržílková Z, Lánská V. Longitudinal trends in cardiovascular mortality and blood pressure levels, prevalence, awareness, treatment, and control of hypertension in the Czech population from 1985 to 2007/2008. J. Hypertens 28, 196-203, 2010.
Guyton AC a Hall JE: Textbook of Medical Physiology, 11th Ed. Elsevier/Saunders, Philadeplhia. 2006.
Ventura O, Taler SJ, Strobeck JE: Hypertension as a hemodynamic disease: The role of impedance cardiography in diagnostic, prognostic, and therapeutic decision making. Am. J. Hypertens. 18, 26S-43S, 2005.
Veselý J: Tlaková diuréza a arteriální hypertenze. Epava, Olomouc, 2002.


{kind=link}
{kind=link}