

pracoviště: Ústav patologické fyziologie LF UP Olomouc, Dětská endokrinologická ambulance Svitavské nemocnice a.s.
Úvod
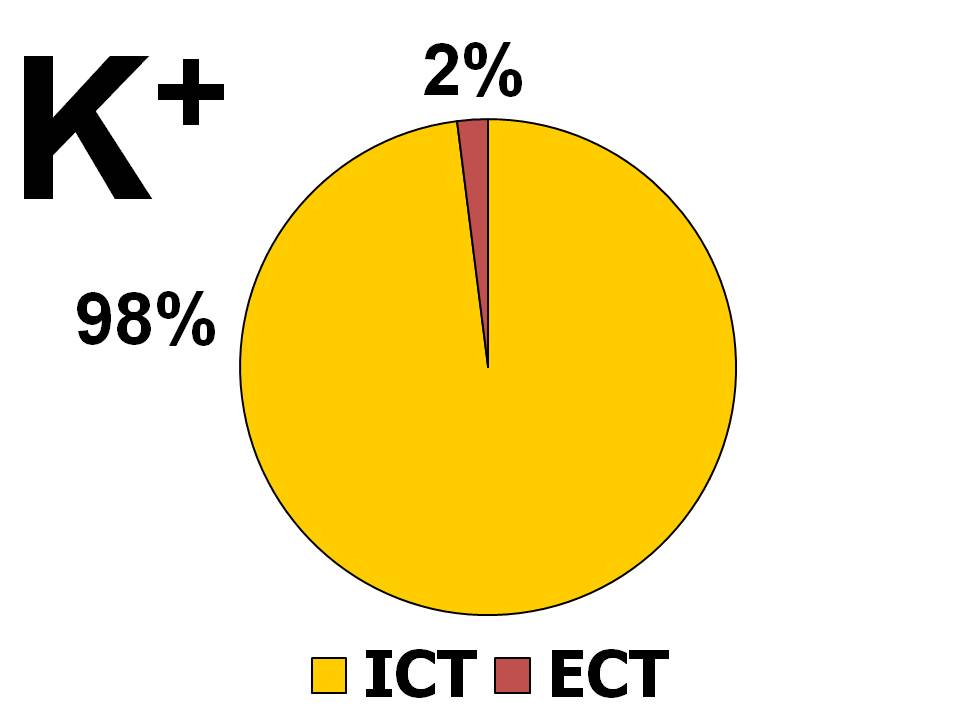
Distribuce draslíku v organismu
Zastoupení iontů draslíku v organismu
- Celých 98 % je uloženo intracelulárně, tzn. uvnitř buněk. Průměrná koncentrace je 140 – 150 mmol K+/l intracelulární tekutiny, přičemž jsou značné rozdíly v obsahu iontů draslíku v závislosti na typu buněk. Tak např. ve svalových buňkách dosahuje koncentrace draselných iontů 160 mmol K+/l, kdežto v erytrocytech jen 45 mmol K+/l. Podle absolutního množství je nejvíce iontů draslíku uloženo ve svalech, a to 60 – 80 % celkových zásob. Svaly, resp. kosterní svaly tak představují významný rezervoár draslíku.
- Pouhá 2 % se nacházejí v extracelulární tekutině, což u dospělého člověka vážícího 70 – 80 kg představuje 70 – 80 mmol/l. Proto také i malá změna v distribuci draslíku mezi ICT a ECT může vést k velkým výkyvům kalémie.
Příjem iontů draslíku ve stravě
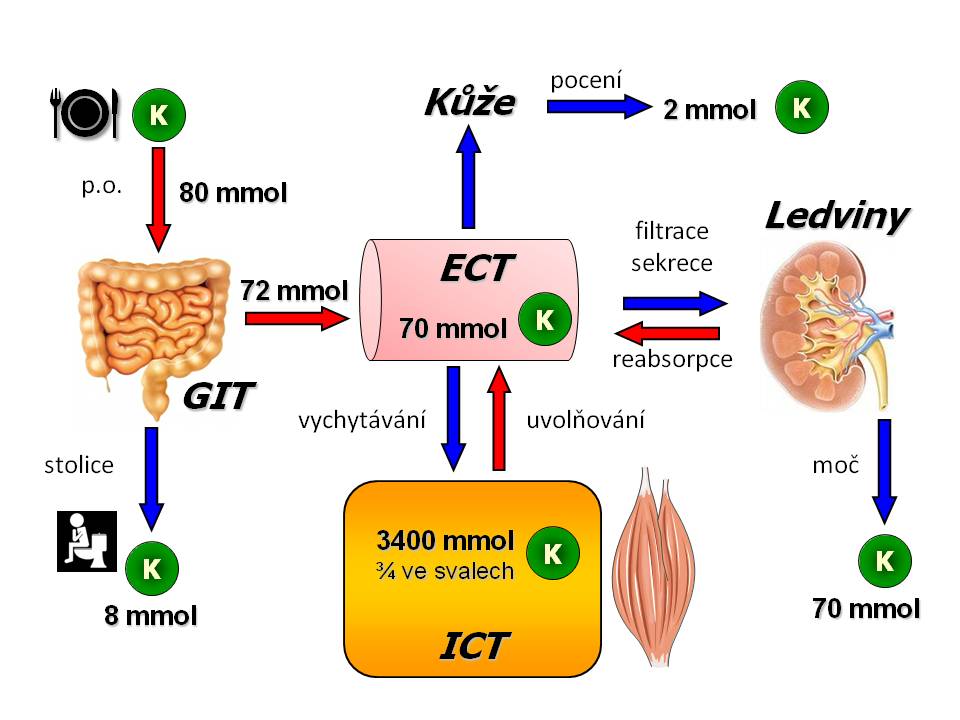
Denní bilance draslíku
Vstřebání a vylučování iontů draslíku v GIT
Svaly jako rezervoár iontů draslíku
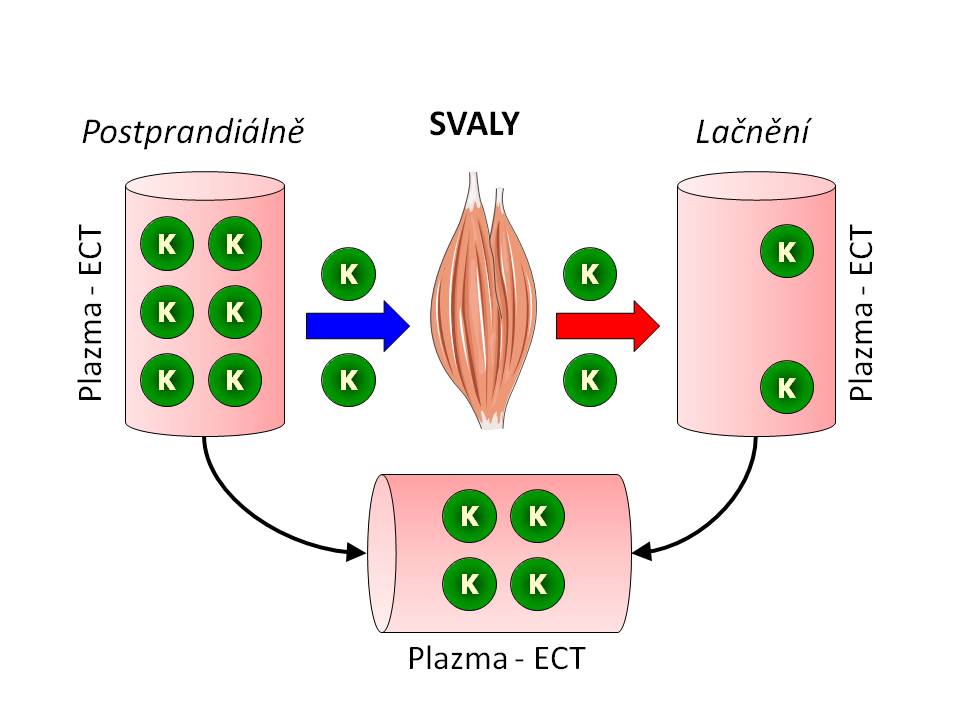
Svaly jako rezervoár draslíku a jejich úloha v pufrování kalemie
Regulace distribuce iontů draslíku mezi extra a intracelulárním oddílem tělesných tekutin
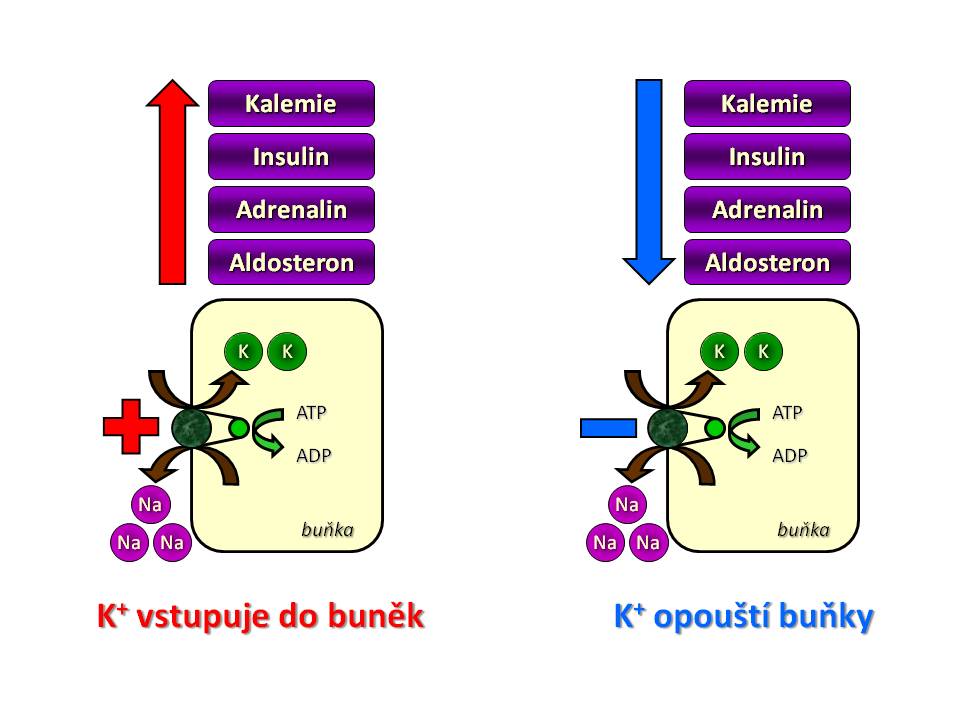
Změna distribuce draslíku mezi ECT a ICT změnou aktivity Na/K-ATPázy
- Kalémie. Stoupající hladina plazmatického kalia sama o sobě zvyšuje aktivitu membránové Na+/K+-ATPázy tím, že zvyšuje dostupnost substrátu, v tomto případě K+, v extracelulárním prostoru.
- Inzulín. Postprandiální vzestup glykémie stimuluje sekreci inzulínu beta buňkami Langerhansových ostrůvků pankreatu a inzulín pak umožňuje nejen vstup glukózy, ale i vstup iontů draslíku do buněk (především do svalových buněk a do hepatocytů), a tím rychle pufruje hladinu plazmatického kalia. Mechanismus, kterým inzulin zvyšuje vstup K+ do buněk, spočívá ve zvýšení aktivity Na+/K+-ATPázy. Inzulín (a) stimuluje antiport Na+/H+, kterým se zvyšuje intracelulární koncentrace iontů sodíku, což substrátově podporuje výměnu Na+ za K+ za spotřeby ATP. (b) Stimuluje syntézu Na+/K+-ATPázy, a tím i celkovou kapacitu transportního systému buněk pro draselné ionty.
- Katecholaminy (především adrenalin) zvyšují vstup iontů draslíku do buněk, protože přímo stimulují aktivitu Na+/K+-ATPázy (b2-receptory), a2-receptory ji však inhibují. Adrenalin také snižuje uvolňování inzulinu.
- Aldosteron mimo svůj hlavní účinek v ledvinách všeobecně zvyšuje vstup iontů draslíku do buněk, a to stejným způsobem jako předchozí faktory, tedy zvýšením aktivity Na+/K+-ATPázy.
Vylučování iontů draslíku močí
- Proximální tubulus: 65 – 70 %;
- Henleova klička – vzestupné („tlusté“) raménko: 25 %;
- Distální tubulus a sběrací kanálek: při depleci draslíku až 9 %.
- Proximální tubulus: 0 %;
- Henleova klička: 0 %;
- Distální tubulus a sběrací kanálek: při normálním příjmu draslíku asi 5 %, při vysokým přívodu draslíku ve stravě až 70 %.
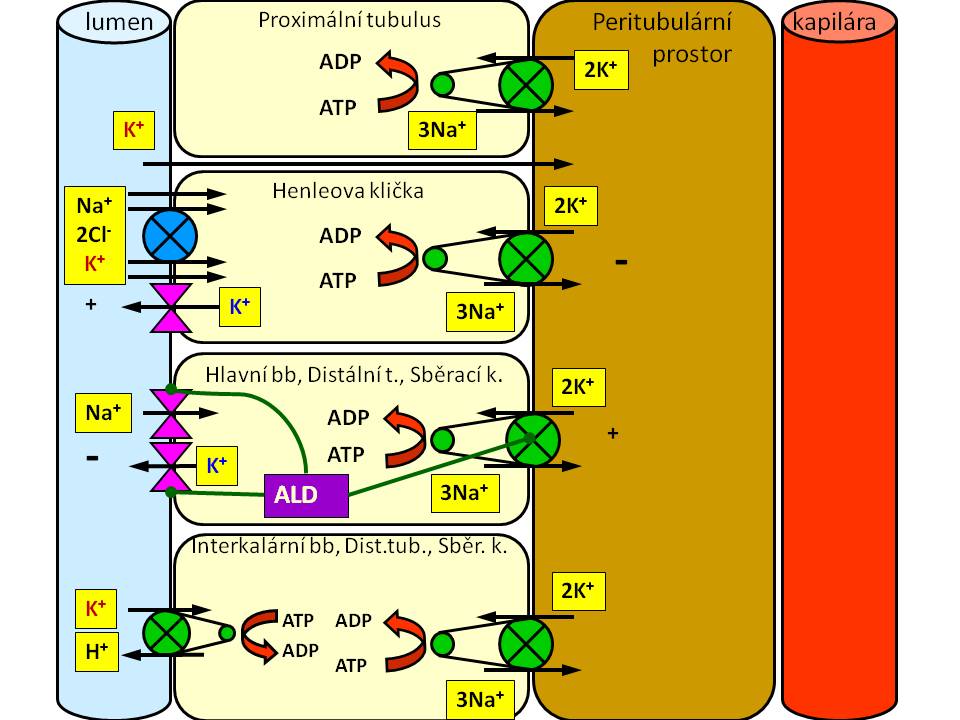
Transportní mechanismy resorpce a sekrece draslíku v ledvinných tubulech
Proximální tubulus. Zde zatím není znám žádný specifický transportér pro K+ lokalizovaný v apikální membráně tubulární buňky. Proto se předpokládá, že zpětná resorpce draslíku zde probíhá difúzí, a to paracelulárními zkraty mezi tubulárními buňkami. Hybnou silou je jednak elektrický gradient, protože lumen proximálního tubulu má ve své druhé části pozitivní náboj, a dále tah rozpustidla, který je dán aktivní resorpcí iontů sodíku, a následně i vody, v proximálním tubulu (viz obrázek).
Henleově klička. V její tlusté vzestupné části, se ionty draslíku resorbují společně se sodnými ionty a chloridy přes NKCC2 (Na-K-Cl-cotransporter). Jde o iontový kanál zajišťující elektroneutrální symport 1Na+1K+: 2Cl-. Hnací silou je elektrochemický gradient pro sodné ionty vytvářené činností Na+/K+-ATPázy v bazolaterální membráně tubulární buňky. Pro činnost tohoto transportéru je potřeba dostatek K+ v luminu tubulu. Proto dochází recirkulaci K+ iontů přes draselný kanál ROMK (renal outer medulla K+ channel, dle systematické klasifikace označovaný jako Kir 1.1) situovaný na luminální membráně. ROMK patří mezi ATP-dependentní K+ kanály. Na bazolaterální straně pak ionty draslíku opouštějí tubulární buňku přes jiný kanál (Kir 6. 1). Část draselných iontů opouští lumen difúzí paracelulárními zkraty po elektrickém gradientu, protože v lumen tubulu je oproti bazolaterálnímu prostoru pozitivní náboj (asi + 15 mV), na jehož tvorbě se podílí Na+/K+-pumpa a již zmíněná recirkulace K+ iontů přes ROMK (viz obrázek).
Distální tubulus a sběrací kanálek. V těchto částech nefronu zajišťují vstřebání iontů draslíku z tubulů tzv. interkalární buňky prostřednictvím K+/H+ antiportu situovanému na luminální membráně a vyžadujícím energii v podobě ATP; za jeden vyloučený H+ se resorbuje jeden K+. Resorpce iontů draslíku v distálním nefronu má za normálních okolností malý význam, uplatňuje se ale při deficitech K+ a také při acidózách, kdy aspoň zčásti koriguje ztráty draslíku, které jsou součástí těchto poruch acidobazické rovnováhy provázejí.
- Nejprve jsou na bazolaterálním pólu ionty draslíku napumpovány z intersticiálního prostoru dovnitř hlavních buněk prostřednictvím Na+/K+-ATPázy. Děje se tak primárním aktivním transportem proti jejich koncentračnímu gradientu, stejně jako jsou z buněk proti koncentračnímu gradientu vypuzovány ionty sodíku; směna probíhá v poměru 3 Na+ ven, 2 K+ dovnitř za spotřeby ATP.
- Následně se ionty draslíku pasivní difúzí přesouvají přes apikální membránu do tubulární tekutiny. Difúze je umožněna příznivým elektrochemickým gradientem a vysokou permeabilitou apikální membrány pro draselné ionty. Ačkoli K+ kanály, umožňující draselným iontů opustit hlavní buňky, jsou lokalizovány jak na jejich apikální, tak na jejich bazolaterální straně, a přesto, že koncentrační gradient pro draselné ionty vzniklý činností Na+/K+-ATPázy je rovněž stejný na obou pólech buňky, ionty draslíku preferenčně unikají přes apikální membránu ze dvou důvodů:
- Lumen tubulů má oproti peritubulárnímu prostoru negativní potenciál (- 35 až – 50 mV). Vzniká přestupem iontů sodíku do buněk; tyto kationty nejsou stejně rychle následovány anionty, takže negativně nabité lumen pak „přitahuje“ pozitivně nabité K+.
- Apikální membrána má vyšší permeabilitu pro K+ než bazální membrána, což je dáno rozdílným typem a vyšší vodivostí kanálů pro K+ v apikální membráně. Principiálně tedy na apikální membráně probíhá směna Na+ za K+. Na+ přestupují z lumina do buněk přes epiteliální kanál sodných iontů (ENaC), kdežto K+ unikají z buněk do lumina tubulů přes ROMK kanály.
- aktivita bazolaterální Na-K-ATPázy;
- elektrochemický gradient pro K+ napříč apikální membránou;
- permeabilita apikální membrány pro K+.
Fyziologická regulace vylučování iontů draslíku ledvinami
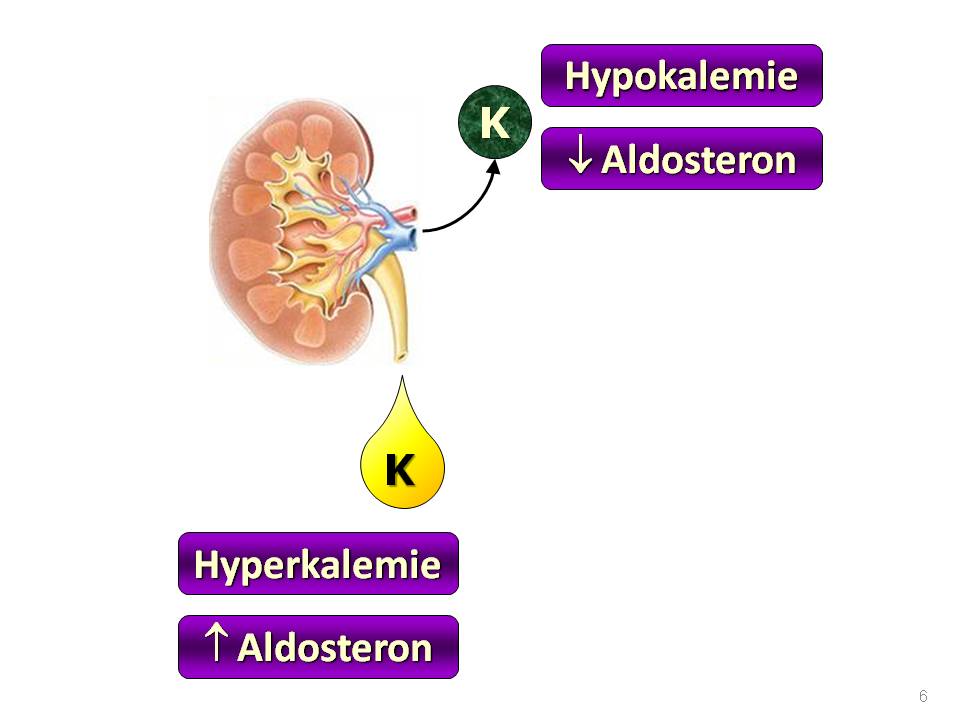
Fyziologické faktory regulující exkreci draslíku ledvinami
Plazmatická koncentrace K+
Hyperkalémie zvyšuje sekreci iontů draslíku v distálních tubulech a sběracích kanálcích z několika důvodů:
- Zvyšuje sekreci aldosteronu – viz níže.
- Stimuluje aktivitu Na+/K+-ATPázy na bazolaterální membráně, protože její činnost přímo závisí na dostatečné nabídce „substrátů“, tzn. dostatku K+ v plazmě stejně jako dostatku Na+ uvnitř buněk (a samozřejmě dostatku ATP). Zvýšením aktivity tohoto transportéru se zvyšuje koncentrace iontů draslíku uvnitř tubulárních buněk, a tím i jeho difúze přes apikální membránu.
- Snižuje koncentrační gradient pro ionty draslíku na bazolaterální membráně, čímž omezuje zpětný únik draslíku z tubulárních buněk přes bazolaterální membránu zpátky do krve a do organismu.
Hypokalémie dělá opak, tedy snižuje sekreci iontů draslíku v distálním nefronu.
Aldosteron
je mineralokortikoid kůry nadledvin, resp. její zona glomerulosa. Právě hyperkalémie je druhým nejvýznamnějším podnětem pro jeho vyplavení po angiotenzinu II. Aldosteron zvyšuje sekreci iontů draslíku hlavními buňkami distálního nefronu z následujících důvodů:
- Zvyšuje kapacitu systému Na+/K+-ATPáz v bazolaterální membráně, protože zvyšuje jejich počet;
- Současně zvyšuje permeabilitu apikální membrány pro ionty draslíku, protože zvyšuje počet ROMK kanálů pro draselné ionty a také ENaC kanálů pro sodné ionty. Právě zvýšená resorpce iontů sodíku pak je hnací silou zvýšené sekrece iontů draslíku. Vztah mezi hladinou draselných iontů v plazmě a sekrecí aldosteronu je exponenciální – změny kalémie ve fyziologických mezích vedou jen k malému vzestupu sekrece aldosteronu, ale zvýšení plazmatické koncentrace iontů draslíku do pásma hyperkalémie vede k velké změně tvorby aldosteronu. Aldosteron je tedy strážcem hyperkalémie.
Zpětná a dopředná vazba v kontrole K+ homeostázy
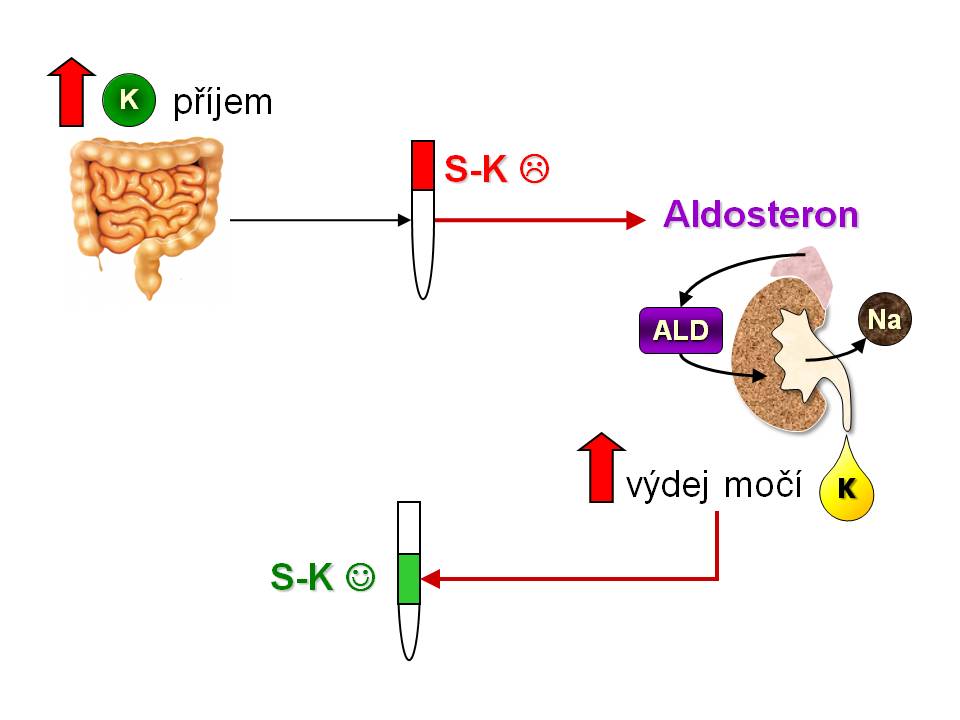
Kontrola homeostázy draslíku zpětnou vazbou
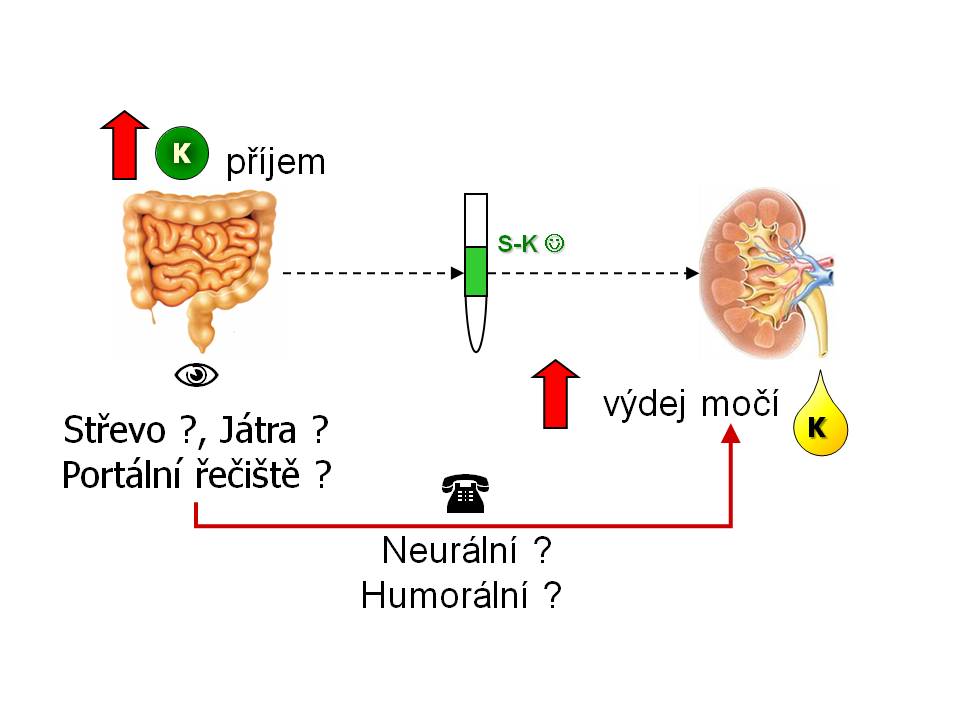
Kontrola homeostázy draslíku dopřednou vazbou
Fyziologický význam iontů draslíku v organismu
K+ a klidový membránový potenciál
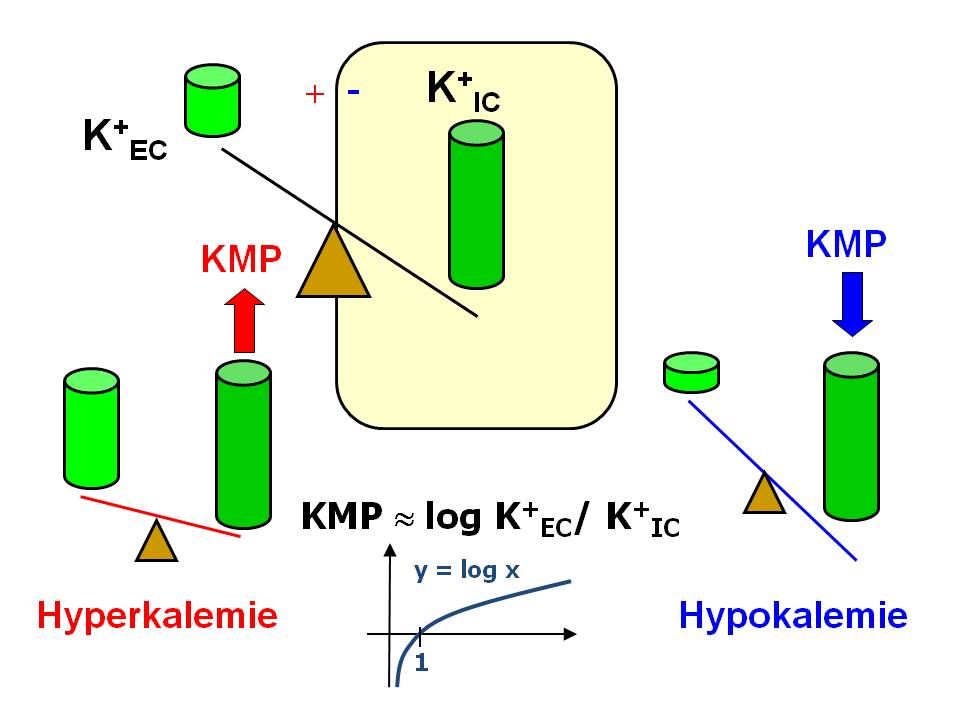
Vliv hyperkalemie a hypokalemie na klidový membránový potenciál
- Přítomností velkých nedifuzibilních aniontů v buňce. Těmito anionty jsou organické fosfáty a proteiny, pro které je plazmatická membrána zcela neprostupná, a tak nemohou sledovat tok malých kationtů unikajících z buňky po koncentračním gradientu, což vede k převaze negativního náboje uvnitř buňky.
- Koncentrační gradient pro malé ionty je udržován aktivní činností několika typů transportérů, z nichž největší význam má membránová Na+/K+-ATPáza, která přesunuje Na+ a K+ proti jejich koncentračním gradientům, tedy Na+ z buněk do ECT, a naopak K+ z ECT do buněk. Tato činnost vyžaduje velkou část ATP produkované buňkami. Aktivita pumpy musí být nepřetržitá, protože oba ionty proudí zpět po svém koncentračním gradientu skrze membránu jednak difúzí, jednak cestou svých specifických iontových kanálů. Vliv Na+/K+-ATPázy na velikost KMP je tedy především nepřímý daný vzniklými koncentračními rozdíly pro jednotlivé ionty. ATPáza nicméně má i přímý elektrogenní vliv, protože ionty Na+ a K+ přesunuje v poměru 3:2, tedy 3 kationty z buňky ven a jen dva kationty do buňky. Tím významně přispívá k negativitě na vnitřní straně plazmatické membrány.
- Rozdílná vodivost (gX) buněčné membrány pro malé ionty. Klidová plazmatická membrána je poměrně vysoce propustná pro K+, méně pro Cl- a velmi málo pro Na+. Vyjádřeno relativně, permeabilita pro K+ je asi 2x větší než pro Cl- a asi 50x vyšší než pro Na+, tzn. relativní vztahy mezi vodivostí pro jednotlivé ionty gK : gN a: gCl jsou v poměru 1.0:0.02:0.5. Tento fakt zohledňuje Goldmannova rovnice KMP = RT/F . ln [(gK . c[K+]EC + gNa . c[Na+]EC + gCl . c[Cl-]IC)/(gK . c[K+]IC + gNa . c[Na+]IC + gCl. c[Cl-]EC). Abychom se mohli vyhnout dosazování absolutních hodnot vodivosti a použili hodnoty relativní, pak si rovnici upravíme do tvaru KMP = rgK/∑g . EK + rgNa/∑g . ENa + rgCl /Σg . ECl , kde EK, ENa, ECl jsou rovnovážné potenciály pro jednotlivé ionty vypočítané z Nernstovy rovnice, a rgK , rgNa , rgCl relativní vodivosti daného iontu v poměru k celkové relativní vodivosti (∑g) pro všechny uvažované ionty. Tedy jestliže určíme rgK jako 1.0, rgNa 0,02 a rgCl 0,5 a ∑g je 1,52 (1,0+0,02+0,5) a použijeme výše uvedené hodnoty rovnovážných potenciálů vypočítané z Nernstovy rovnice (EK -90 mV, ENa +70 mV a ECl -80 mV), pak výsledný KMP je -85 mV.
K+ a akční potenciál vzrušivých tkání

Vliv hyperkalemie a hypokalemie na akční potenciál kardiomyocytů
1. Depolarizace. Po dosažení prahové úrovně dojde otevřením napěťově řízených natriových kanálů k rychlému nárůstu permeability membrány pro sodné ionty. Kationty Na+ proudí do buňky ve směru chemického i elektrického gradientu přinášejíce s sebou pozitivní náboj, takže negativita vnitřku buňky se rychle snižuje, až nakonec dojde k „překmitu“ do pozitivních hodnot. Na vrcholu AP je elektrický potenciál uvnitř buňky o + 30 až + 40 mV vyšší (pozitivnější) než vně buňky.
2. Repolarizace. Přestřelení membránového potenciálu natriové kanály zase rychle inaktivuje a proud sodných iontů směřujících pozitivní náboj do buňky ustává. Již během depolarizace stoupá vodivost membrány pro ionty draslíku, a to díky otevírání napěťově řízených kaliových kanálů (jde o jiný typ K-kanálů, než jaký se podílí na udržování KMP). Jimi proudí kationty K+ z buňky ven a odnášejí s sebou pozitivní náboj. Ovšem jejich otevírání je pozvolnější, a tak vrchol vodivosti pro ionty draslíku přichází později než pro ionty sodíku. V momentě, kdy se sodné kanály uzavřou, začne se díky proudu draselných iontů polarita membrány vracet zpět do negativních hodnot, dokud buňka neobnoví svůj KMP. Kaliové kanály, odpovědné za tuto fázi AP, se s prohlubující se repolarizací postupně uzavírají.
1. Depolarizace. Probíhá obdobně jakO u buněk ostatních svalů nebo u nervových buněk. Podkladem je přechodné zvýšení permeability pro ionty sodíku s následným depolarizujícím proudem kationtů Na+ do cytoplazmy kardiomyocytů a s překmitem membránového napětí do pozitivních hodnot (+ 20 mV).
2. Plateau. Po uzavření sodných kanálů se vlivem již otevřených draselných kanálů o něco sníží pozitivita membránového potenciálu (na hodnoty lehce nad 0 mV), ale pak zůstává po značnou dobu neměnná (200 – 300 ms). Příčinou jsou dva protichůdné iontové proudy - depolarizující proud vápenatých iontů, který do buňky přináší Ca2+ ionty po jejich koncentračním gradientu, a proti němu působící repolarizující proud draselných iontů, kterým K+ ionty unikají z buňky ven.
3. Repolarizace. Po uzavření kalciových kanálů končí fáze plateau a nastává repolarizace. Otevřené zůstávají pouze kanály pro draselné ionty. Repolarizující proud K+ vynáší pozitivní náboj z nitra kardiomyocytů ven, dokud nedojde k obnově KMP.
K+ a intracelulární osmolarita
K+ a acidobazická rovnováha
Poruchy homeostázy draselných iontů

Klasifikace poruch bilance draslíku
Deficit K+. Jde o poruchu bilance iontů draslíku, kdy výdej převyšuje jejich příjem.
Nadbytek K+. Jde o poruchu bilance iontů draslíku, kdy příjem převyšuje jejich výdej.
Hypokalémie. Jde o snížení koncentrace iontů draslíku v plazmě pod dolní hranici normy, tedy pod 3,7 mmol/l.
Hyperkalémie. Jde o zvýšení koncentrace iontů draslíku v plazmě nad horní hranici normy, tedy nad 5,3 mmol/l.
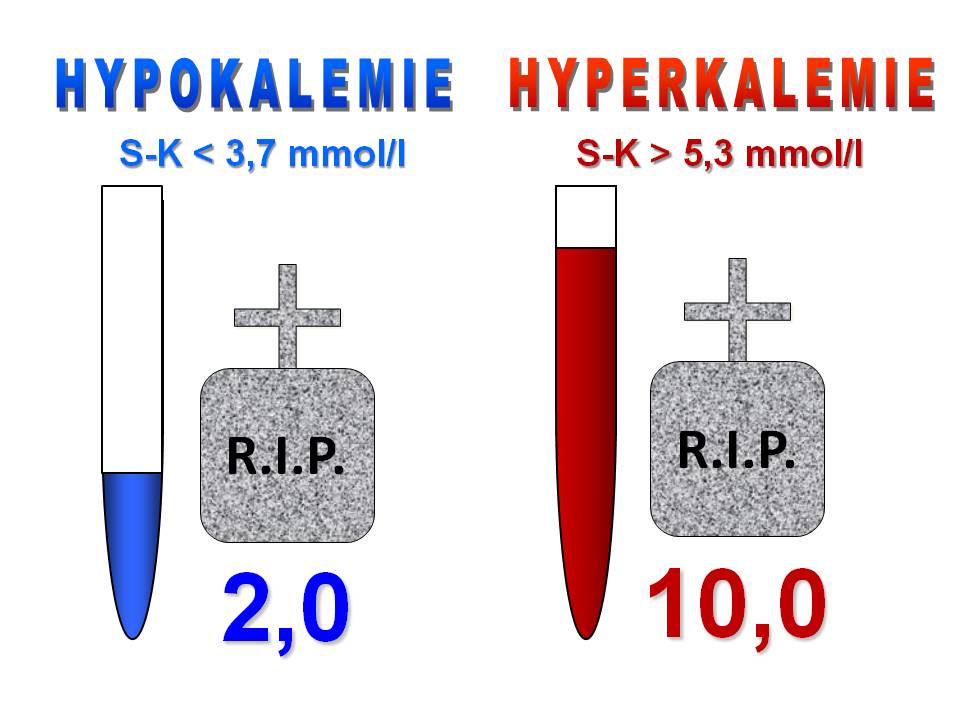
Klasifikace poruch plazmatické koncentrace draslíku
Klasifikace hypokalémie
Etiologie hypokalémie

Příčiny hypokalemie
- Nízký přísun iontů draslíku do organismu;
- Zvýšené ztráty iontů draslíku z organismu;
- Poruchu distribuce iontů draslíku v rámci různých oddílů organismu.
Nízký přísun iontů draslíku do organismu
Ztráty iontů draslíku cestou GIT
Ztráty iontů draslíku potem
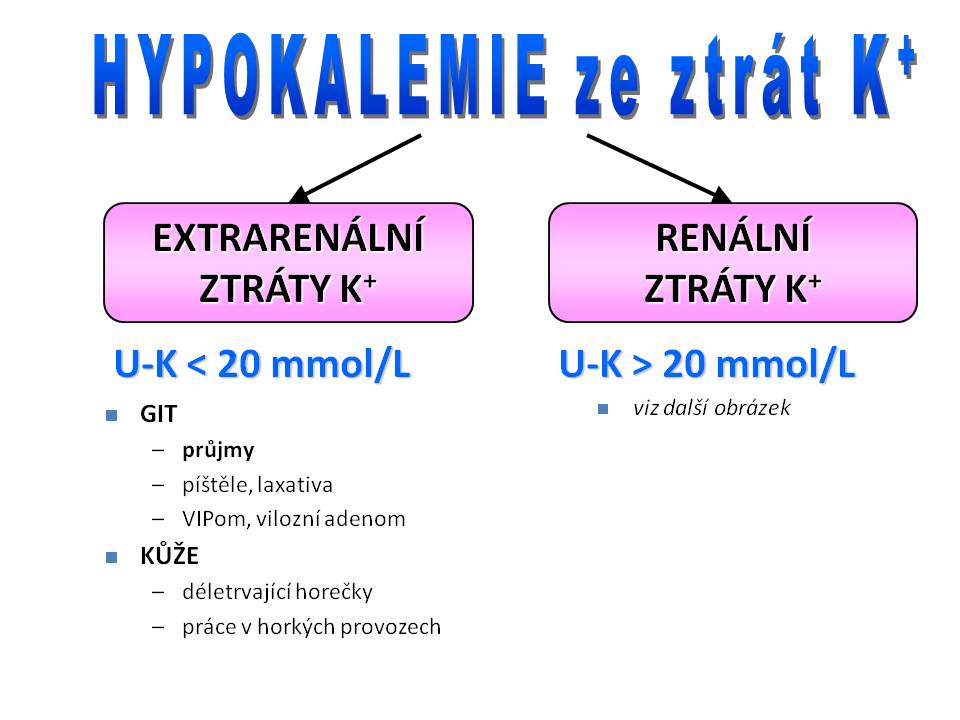
Příčiny ztrát draslíku z organismu
Ztráty iontů draslíku ledvinami
Jsou nejčastější příčinou hypokalémie a deplece kalia. Vzhledem k rozsáhlé diferenciální diagnostice těchto stavů je namístě jejich členění dle stavu acidobazické rovnováhy (ABR):
Renální ztráty iontů draslíku bez významnější poruchy ABR. Sem jako příčinu můžeme zařadit polyurickou fázi akutní tubulární nekrózy nebo zvýšenou diurézu po odstranění obstruktivní uropatie – příčinou zde je zvýšený přísun iontů sodíku do distálního tubulu, a tím jeho zvýšená resorpce výměnou za ionty draslíku. Velké dávky penicilinu rovněž zvyšují kaliurézu, protože špatně resorbovatelný aniont penicilinu v moči zvyšuje negativitu intraluminálního potenciálu, a tím i exkreci iontů draslíku hlavními buňkami. Z terapeutického hlediska je u hypokalémií vždy nutno došetřit homeostázu iontů hořčíku, protože hypomagnezémie, resp. deplece iontů hořčíku, ne zcela jasným mechanismem rovněž zvyšuje ztráty draselných iontů ledvinami. Substituce draselnými ionty pak je neúčinná, pokud nebudou současně doplněny i zásoby iontů hořčíku. K deficitu Mg2+, a tím i deficitu K+, vedou některé léky jako např. cisplatina, amfotericin B aj.
Renální ztráty iontů draslíku spojené s metabolickou acidózou. Typickým příkladem z této skupiny je renální tubulární acidóza (RTA). Příčiny RTA jsou buď vrozené, nebo získané, přičemž rozeznáváme:
- Proximální RTA, kdy jde o poruchu resorpce bikarbonátů v proximálním tubulu. Neresorbované HCO3- v distálním tubulu prohlubují negativitu intraluminálního potenciálu, a tím zvyšují sekreci K+.
- Distální RTA, kdy je porušena sekrece protonů do moči buňkami distálního tubulu - na luminální membráně tubulární buňky vázne protitransport H+ (ven) za K+ (dovnitř). Obdobná situace jako u proximální RTA je při užití inhibitorů karbonátdehydratázy (karboanhydrázy) (např. acetazolamidu).
- Velkou deplecí iontů draslíku je rovněž provázena diabetická ketoacidóza, kde se na ztrátách kalia močí podílí především osmotická diuréza.
- Hypokalémie z renálních ztrát se také vyskytuje při čichání toluenu, který se v organismu metabolizuje na kyselinu hippurovou, jejíž aniont hippurát je z tubulů neresorbovatelný, a tím zvyšuje vylučování K+ v distálním nefronu.
Renální ztráty iontů draslíku spojené s metabolickou alkalózou a s normálním anebo nižším krevním tlakem. Nejčastější příčinou hypokalémie je v klinické praxi léčba diuretiky (vyjma kalium šetřících diuretik). Jak kličková, tak thiazidová diuretika zablokováním zpětné resorpce iontů sodíku ve vyšších etážích nefronu zvyšují jejich přísun do druhé části distálních tubulů a do kortikální části sběracích kanálků, kde je příliv Na+ částečně zpětně resorbován výměnou za K+. Volumová deplece navíc vede k sekundárnímu hyperaldosteronismu, a tím i potenciaci sekrece K+ do lumen tubulů v této části nefronu. Větší kaliuretický efekt vykazují thiazidová diuretika, což je částečně vysvětlitelné jejich delším farmakologickým poločasem.
Stejným mechanismem je vysvětlitelná hypokalémie provázející dvě vzácné vrozené tubulární poruchy, a to
- Bartterův syndrom, kdy jde o defekt resorpce soli v tlusté části Henleovy kličky, jehož příčinou je buď (a) mutace NKKC2 kontransportéru pro elektroneutrální přesun Na+K+2Cl- na luminální membráně, nebo (b) mutace ROMK, tedy kanálu, kterým ionty draslíku recirkulují na luminální membráně, nebo (c) mutace ClC-K, tedy chloridového kanálu na bazolaterální membráně, kterým aniont Cl- opouští tubulární buňku.
- Gitelmanův syndrom, kdy jde o poruchu resorpce NaCl ve stočené části distálního tubulu, jehož příčinou je mutace NCC, tedy kanálu zajišťujícího kotransport Na+ a Cl- z lumina do buňky. Pro oba syndromy je společná přítomnost hypokalémie, hypochloremické metabolické acidózy, hypomagnesémie a normálního nebo nižšího krevního tlaku, a to přes vysokou hladinu aldosteronu a reninu. Rozdíl je ve vylučování iontů vápníku - u Bartterova syndromu je hyperkalciurie, kdežto u Gitelmanova syndromu hypokalciurie.
Do této podskupiny můžeme dále zařadit všechny situace spojené s absolutní nebo relativní volumovou deplecí, které vedou k aktivaci systému renin-angiotenzin II a k sekundárnímu hyperaldosteronismu, a tím ke zvýšení renálního vylučování draslíku. Jde zejména o krvácení, dehydratace, popáleniny, srdeční selhání, jaterní selhání, nefrotický sy atd. Při zvracení se kromě mechanismu sekundárního hyperaldosteronismu uplatňuje také deficit chloridů a metabolická alkalóza – její kompenzace v ledvinách vede k hyperbikarbonáturii, která prohlubuje negativitu luminálního potenciálu v distálním nefronu, a tím podporuje vylučování iontů draslíku (přímé ztráty draselných iontů zvratky nejsou velké, protože jeho koncentrace v žaludeční šťávě je velmi nízká).
-
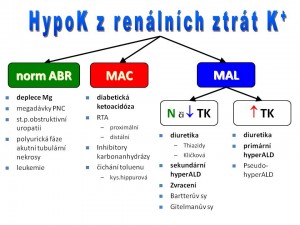
Příčiny renálních ztrát draslíku
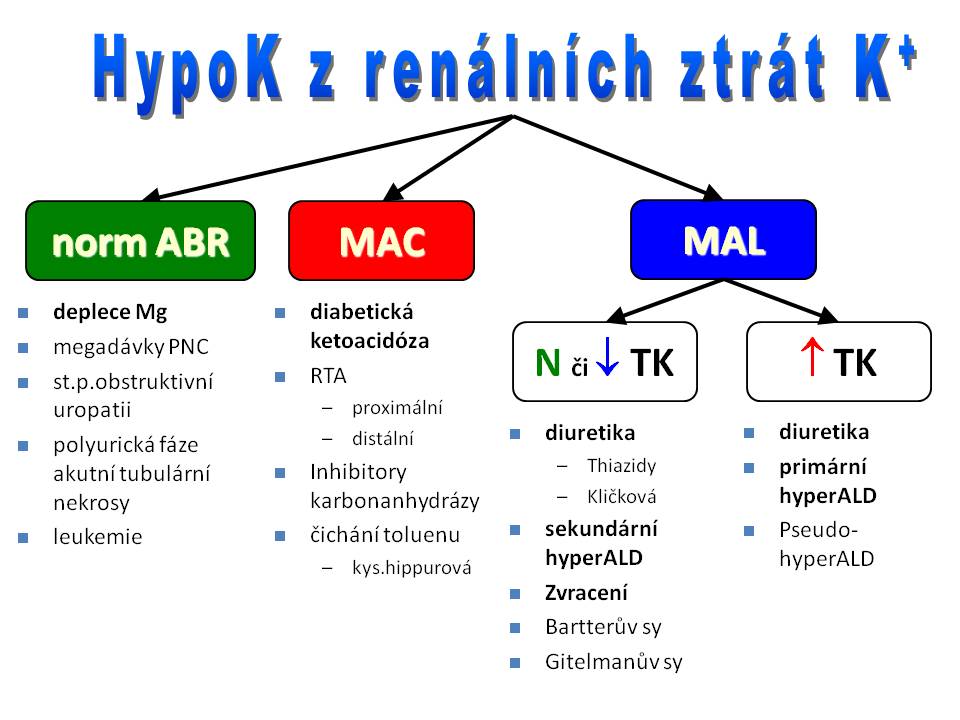
Renální ztráty iontů draslíku spojené s metabolickou alkalózou a s vysokým krevním tlakem.
Samozřejmě i v této podskupině můžeme nalézt pacienty léčené diuretiky. Zejména jde o pacienty trpící hypertenzí, jejichž krevní tlak se z nějakého důvodu nepodařilo dostat pod kontrolu, anebo o pacienty s maligní akcelerací hypertenze. Po vyloučení této možnosti směřuje diferenciální diagnostika cestou zhodnocení reninu (plazmatické reninové aktivity) a hladiny aldosteronu.
- Máme-li nízký renin a vysoký aldosteron jedná se o primární hyperaldosteronismus (Connův syndrom), jehož příčinou je hyperplázie nebo hormonálně aktivní adenom (vzácně karcinom) kůry nadledvin. Nadprodukce aldosteronu také kromě zvýšené sekrece iontů draslíku ledvinami zvyšuje sekreci protonů hlavními buňkami, a tím vede k metabolické alkalóze, která pak prohlubuje hypokalémii přesunem iontů draslíku do buněk výměnou za H+. Raritně jde o hyperaldosteronismus supresibilní glukokortikoidy, což je autosomálně dominantně dědičná porucha steroidogeneze v kůře nadledvin, kdy dojde k fúzi genu aldosteronsyntázy a regulační sekvence genu 11β-hydroxylázy, čímž se tvorba a sekrece aldosteronu přesune z dominantní kontroly angiotenzinem II pod dominantní kontrolu ACTH. Stav je příznivě ovlivnitelný podáním glukokortikoidů, které zpětnovazebně ztlumí sekreci ACTH, a tím i aldosteronu.
- Je-li vysoký renin a vysoký aldosteron, může být příčinou renovaskulární obstrukce, nebo reninom – tedy nádor secernující renin; obé vede k rozvoji hypokalémie a volumové hypertenze.
- Při výsledku nízký renin a nízký aldosteron je potřeba pátrat po jiném hormonu nebo faktoru, které mají mineralokortikoidní účinek. Na prvním místě může jít o Cushingův syndrom, ať už periferní, centrální anebo iatrogenní. Příčinou je zde nadbytek endogenního kortizolu nebo exogenních kortikoidů, které jsou sice slabé mineralokortikoidy, ale při vysokých hladinách se vážou na mineralokortikoidní receptory. Další příčinou může být pseudohyperaldosteronismus, kam řadíme následující nosologické jednotky: (1) Vzácnější formu kongenitální adrenální hyperplázie (CAH), při které je mutován gen pro 11-β-hydroxylázu. Výsledkem je nedostatečná syntéza kortizolu, která zvyšuje sekreci ACTH. Ten následně stimuluje tvorbu nadledvinových androgenů. Zároveň se před enzymovým blokem hromadí 11-deoxykortikosteron, což je mineralokortikoidně působící prekurzor aldosteronu. Výsledkem je virilizace genitálu u děvčátek, předčasná puberta u chlapců a u obou pohlaví hypertenze s hypokalémií. (2) Liddleův syndrom, což je autozomálně dominantně dědičná mutace epiteliálního sodíkového kanálu (ENaC) hlavních buněk distálních tubulů a sběracích kanálků. Jde o mutaci aktivační, takže dochází k nekontrolovaně vysoké resorpci iontů sodíku bez vlivu aldosteronu, a následně k rozvoji hypertenze. (3) Intoxikaci lékořicí, jejíž extrakty se zejména dříve používaly v lidové medicíně pro své expektorační, antiflogistické a gastroprotektivní účinky. Lékořice obsahuje kyselinu glycyrrhiziovou, která blokuje 11-β-hydroxysteroiddehydrogenázu 2. typu (11-β -HSD 2). Tento enzym fyziologicky brání mineralokortikoidnímu účinku glukokortikoidů na ledviny tím, že přeměňuje kortizol na neaktivní metabolit kortizon. Při jeho deficitu pak i běžné, nezvýšené hladiny kortizolu mají mineralokortikoidní efekt (z jiného úhlu pohledu tedy vzniká hypertenze u Cushingova syndromu následkem nedostatečné kapacity tohoto enzymu, která nezvládá příval vysokých hladin kortizolu v ledvinách).
Porucha distribuce iontů draslíku
Inzulín. Akutní léčba inzulínem vede ke vstupu nejen glukózy, ale i iontů draslíku do buněk. Každý obrat katabolismu k anabolismu je provázen přesunem draselných iontů (stejně jako iontů hořčíku a fosforečnanů) do buněk. Na tuto skutečnost je třeba myslet zejména při léčbě diabetické ketoacidózy, protože ta je vždy provázena výrazným deficitem iontů draslíku v buňkách, ale samotná kalémie bývá díky přítomné metabolické acidóze před zahájením léčby normální, nebo dokonce lehce zvýšená, a tím „maskuje“ přítomný deficit (téma Patofyziologie a klinická fyziologie diabetes mellitus a téma Acidobazická rovnováha a její poruchy).
Hyperaldosteronismus. Aldosteron vede k hypokalémii především svým kaliuretickým účinkem v ledvinách, ale v menší míře se na vzniku hypokalémie při hyperaldosteronismu podílí i přesun iontů draslíku do buněk stimulací tvorby Na+/K+-ATPáz.
Metabolická alkalóza. Podporuje výměnu H+ iontů vystupujících z buněk za kationty K+ vstupující do buněk.
Rychlá buněčná proliferace. Pěkným příkladem je léčba perniciózní anémie, kdy po podání vitaminu B12 dojde k restartu erytropoézy a k rychlému vychytání iontů draslíku z plazmy nově se formujícími erytrocyty v kostní dřeni.
Jiným příkladem je tzv. pseudohypokalémie u pacientů s akutní myeloidní leukémií a extrémní leukocytózou v periferní krvi, kdy ve vzorku krve ponechaném delší čas při pokojové teplotě dojde k vychytání iontů draslíku leukemickými buňkami. Situaci lze předejít buď rychlou separací plazmy od celulární složky krve anebo uskladněním vzorku krve v chladu.
Transfúze předtím zmražené krve. Stejně jako v předešlém případě masa buněk, v tomto případě transfundovaných erytrocytů, vychytává ionty draslíku z plazmy.
Tyreotoxická hypokalemická paralýza. Tyroidní hormony mimo jiné svými účinky rovněž zvyšují aktivitu membránových Na+/K+-ATPáz, a tím i vstup iontů draslíku do buněk. Hypokalemické paralýzy v asociaci s tyreotoxikózou, tj. nadprodukcí tyroidních hormonů, se častěji vyskytují u asijského etnika.
Familiální hypokalemická periodická paralýza je vzácná autosomálně dědičná choroba charakterizovaná náhlými atakami generalizované svalové paralýzy na podkladě distribuční hypokalémie. Příčinou je mutace napěťově řízeného kalciového kanálu kosterního svalstva. Spouštěcím faktorem může být stres, intenzivní fyzické cvičení (katecholaminy) anebo strava s vysokým obsahem cukrů (inzulín). Častěji se objevuje v noci nebo v brzkém ránu. Obrna odeznívá spontánně během 6 až 24 hodin.
β2-Adrenergní agonisté. Mohou být původu endogenního, jak je tomu při stresové odpovědi, kdy se vyplaví katecholaminy adrenalin a noradrenalin, nebo původu exogenního – např. při léčbě astmatického záchvatu bronchodilatancii ze skupiny β2-sympatomimetik, jako je např. salbutamol, kdy může dojít k poklesu kalémie i o více jak 0,5 – 1,0 mmol/l. Ke stejnému efektu může vést i předávkování dalšími léky ze skupiny β2-adrenergních agonistů. Příkladem je použití tokolytik využívaných v porodnictví ke ztlumení děložních kontrakcí, nebo užívání pseudoefedrinu, který je součástí některých léků na „nachlazení“, protože vede k dekongesci sliznic (přes α1-adrenergní receptory), je také oblíben na drogové scéně k výrobě pervitinu.
Xantiny, jako jsou teofylin nebo kofein, zvyšují aktivitu Na+/K+-ATPázy nepřímo přes inhibici fosfodiesterázy (čímž inhibují degradaci cAMP) a také zvyšují uvolňování katecholaminů.
Další léky. Blokátory kalciových kanálů v terapeutických dávkách nemají na plazmatickou koncentraci draselných iontů prakticky žádný vliv, ale při předávkování nebo otravách verapamilem byly popsány těžké hypokalémie. Soli baria blokují kaliové kanály, a tím blokují únik iontů draslíku z buněk. Síran barnatý používaný v radiologii jako kontrastní látka ale nevede k otravě, protože se nevstřebává z GIT. Lék chloroquin používaný v léčbě malarie také blokuje výtokové kaliové kanály. Hypokalémie způsobené přesunem iontů draslíku do buněk byly také popsány při otravách psychofarmakem risperidonem.
Příznaky hypokalémie
- (a) rychlost rozvoje hypokalémie,
- (b) přítomnost, nebo nepřítomnost přidružených, zejména kardiálních onemocnění (ICHS, st. p. IM, srdeční selhání, hypertrofické srdce, vrozený syndrom dlouhého QT atp.),
- (c) případně současné užívání léků, jejichž toxicita stoupá při hypokalémii (digoxin). Nejvýznamnější symptomatologie souvisí s postižením činnosti srdce a ostatních svalů.
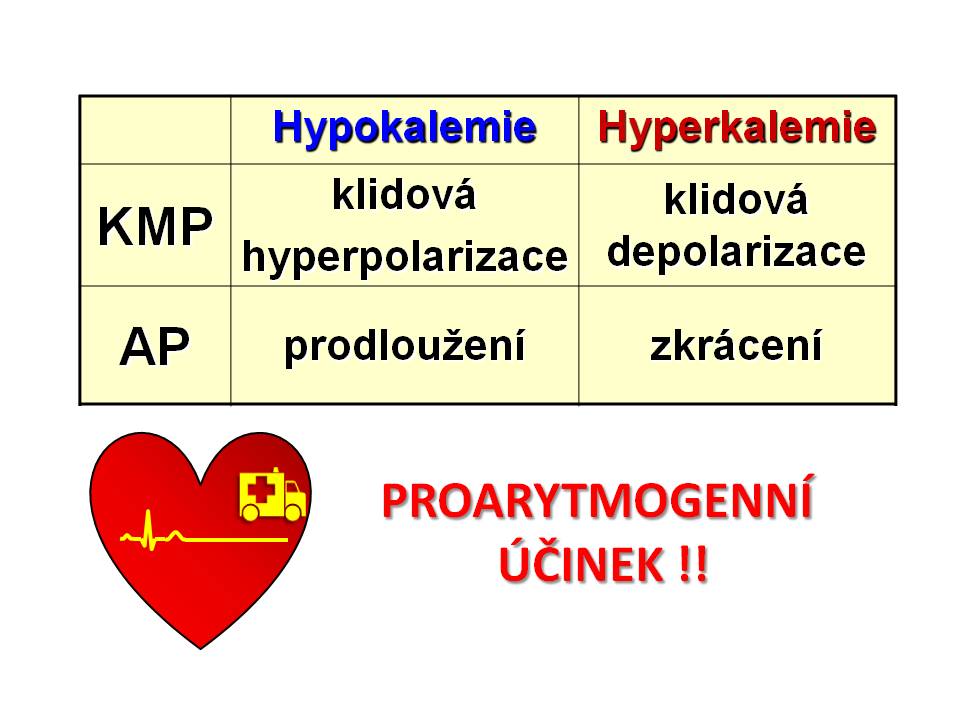
Souhrn patogeneze proarytmogenního účinku hypokalemie a hyperkalemie
SRDCE. Hypokalémie působí proarytmogenně, a to zejména ve skupině kardiologických pacientů. Nejrizikovější skupinu představují pacienti s arteriální hypertenzí a srdečním selháním léčení diuretiky. Hypokalémie představuje nezávislý rizikový faktor náhlé kardiální smrti následkem elektrické instability. Klesající hladina plazmatického kalia inverzně koreluje se stoupajícím rizikem život ohrožujících arytmií včetně fibrilace komor. Vliv hypokalémie na elektrickou stabilitu srdce je komplexní a zahrnuje: (a) Přímý proarytmogenní efekt na myokard; (b) Nepřímý proarytmogenní efekt, když hypokalémie zesiluje proarytmogenní účinek léků užívaných u srdečních onemocnění (digoxin, antiarytmika, katecholaminy); (c) Nepřímý proarytmogenní efekt, když hypokalémie zvyšuje pravděpodobnost vzniku srdečních arytmií u strukturních poškození srdce (ischémie, hypertrofie, dilatace). Podstata přímého proarytmogenního účinku hypokalémie na srdce spočívá ve změnách membránového potenciálu jak pracovního, tak převodního myokardu. Z elektrofyziologického hlediska hypokalémie prodlužuje třetí fázi AP a prohlubuje negativitu KMP. Následkem toho dochází k následujícím změnám
- prodlužení komorové repolarizace,
- zpomalení vedení vzruchu,
- abnormální komorová automacie,
- spontánně spouštěná aktivita,
- vznik krouživých (reentry) vzruchů.
Prodloužená komorová repolarizace. Jak víme z předchozího výkladu, je 3. fáze akčního potenciálu, tj. repolarizace buněk pracovního myokardu, zprostředkována výstupem iontů draslíku z buňky, čímž je vynášen pozitivní náboj a dochází k obnově negativního potenciálu na vnitřní straně buněčné membrány kardiomyocytů. Tento tok iontů draslíku je zprostředkován cestou několika typů kaliových kanálů. Hypokalémie snižuje vodivost membrány pro ionty draslík předčasnou inaktivací jednoho z proudů draselných iontů zajišťujících repolarizaci. Výsledkem je protrahovaná komorová repolarizace, která se na EKG projeví jako prodloužená, oploštělá vlna T, bifazická vlna T, popř. se objeví vlny U, a celkově se prodlouží interval QT.

EKG projevy hypokalemie
Pomalé vedení vzruchu. Výše KMP je závislá na logaritmu poměru koncentrací iontů draslíku vně a uvnitř buněk (KMP ~ log c[K]EC / c[K]IC), a proto hypokalémie vede k prohloubení negativity KMP. Následkem hyperpolarizace je snížená excitabilita, kdy je k vyvolání AP zapotřebí vzruchu o větší intenzitě, protože je větší rozdíl mezi hodnotou KMP a prahem pro otevření rychlých natriových kanálů. Snížená excitabilita pak zpomaluje rychlost vedení, a to se při těžší hypokalémii projeví prodloužením PQ intervalu a rozšířením QRS komplexu na EKG.
Abnormální komorová automacie. Hypokalémie v buňkách Purkyňových vláken zvyšuje rychlost spontánní diastolické depolarizace. Tím se tato část převodního systému může stát zdrojem aktivní heterotopní poruchy automacie a dát vznik předčasným stahům a rytmům – tedy komorovým extrasystolám a tachykardiím.
Spouštěná aktivita. Riziko protrahované repolarizace spočívá ve vzniku časných následných depolarizací. Jde o spontánní oscilace membránového napětí, které narušují průběh repolarizace. Jestliže následné depolarizace dosáhnou prahu pro vznik AP, mohou vést k samovolnému spuštění nové vlny depolarizace. Mluvíme o tzv. spouštěné aktivitě,. Jejím výsledkem může být komorová extrasystola, ale také setrvalá komorová tachykardie včetně život ohrožující arytmie typu torsade de pointes.
Hypokalémie kromě časných následných depolarizací indukuje i vznik pozdních následných depolarizací, kdy spontánní oscilace napětí přicházejí na konci anebo těsně po skončení repolarizace. Souvisejí s přetížením kardiomyocytů ionty vápníku. Proč právě vápenatými ionty? Hypokalémie tlumí aktivitu Na+/K+-ATPázy. To vede k vzestupu intracelulární koncentrace sodných iontů, které stimulují Na+/Ca2+ výměník. Protitrasnport Na+/Ca2+ není elektroneutrální. Převaha kladného náboje je na straně vápenatých iontů. Ty pak depolarizují membránu s rizikem spuštění dalšího AP.
Reentry tachykardie. Riziko komorových arytmií je o to větší, že prodloužení repolarizace není provázeno proporcionálním prodloužením efektivní refrakterní fáze. Studie dokonce ukazují, že efektivní refrakterní fáze je zkrácena, a tím se otevírá časové i prostorové okno pro reexcitaci ještě před skončením repolarizace. Tím vyvstává riziko vzniku krouživých vzruchů a na jejich podkladě pak reentry tachyarytmií. Různé zastoupení kaliových kanálů v různých částech myokardu způsobuje heterogenní odpověď myokardu na hypokalémii. To může podporovat vznik jednosměrného bloku v určité oblasti myokardu, a to je jednou z podmínek vzniku reentry tachyarytmií – vč. smrtící fibrilace komor. K elektrické nehomogenitě myokardu, a tím i hrozbě uzavření reentry okruhu, rovněž příspívá pomalá excitační vlna.
OBĚH. Nedostatečný přísun iontů draslíku do organismu vede k rozvoji arteriální hypertenze, a to pravděpodobně mechanismem zvýšené retence iontů sodíku, a tím i vody, v ledvinách při depleci draslíku. Draselné ionty jsou samy osobě slabé antihypertenzivum. Důležitější pro výši krevního tlaku není jejich absolutní množství ve stravě, ale jejich poměr k současně přijímaným „prohypertenzním“ iontům sodíku. Fylogenetickým vývojem je člověk nastaven na poměr Na+ ku K+ ve stravě 1 : 8-9, jenže v současnosti v zemích typu Česka je poměr obrácený – 2 až 4 : 1, tedy takový, na jaký naše geny a fyziologické regulace nejsou evolučně připraveny. Nedostatek iontů draslíku zvyšuje citlivost arteriálního tlaku na příjem soli, a tak množství NaCl, které by při normálním zásobení těla draselnými ionty nevyvolalo hypertenzi, ji může vyvolat při sníženém přivodu K+ – při depleci draselných iontů stačí k vyvolání hypertenze menší množství NaCl.
KOSTERNÍ SVALY. Hypokalémie vede k prohloubení negativity KMP, což znesnadňuje zahájení AP a tím i svalové kontrakce. Pacienti si stěžují na svalovou slabost, snadnou unavitelnost, myalgie. Při těžké hypokalémii dochází k ascendentně postupující chabé obrně kosterního svalstva. Při velmi těžké hypokalémii (pod 2,0 mmol/l) hrozí smrt paralýzou respiračních svalů. Těžká hypokalémie může také způsobit rhabdomyolýzu během fyzického cvičení, a to následkem nedostatečně zvýšené perfúze pracujícího svalu krví. Vysvětlení je následující - za fyziologických okolností dochází při svalové činnosti k uvolnění iontů draslíku z buněk, což v pracujícím svalu spouští vazodilataci. Při hypokalémii je tato schopnost lokální autoregulační vazodilatace snížená, postižený sval trpí ischémií a může se rozpadnout. Rhabdomyolýza pak může vést k myoglobinurii anebo až k akutnímu renálnímu selhání.
HLADKÉ SVALSTVO GIT. Stejně jako u svalů kosterních vede hypokalémie i v případě hladkého svalstva zažívacího traktu ke slabosti plynoucí z hyperpolarizace plazmatické membrány myocytů. To se projeví jako zácpa. Při těžké hypokalémii může stav progredovat až do paralytického ileu, charakterizovaného úplnou zástavou střevní peristaltiky, vzedmutím břicha, zvracením, dehydratací a deplecí minerálů s rizikem rozvoje šokového stavu a celkové infekce (téma Střevní neprůchodnost (ileus) a pseudoileus).
ACIDOBAZICKÁ ROVNOVÁHA. Hypokalémie v proximálním tubulu stimuluje reabsorpci bikarbonátů, v kortikální části sběracích kanálků stimuluje H+/K+-ATPázu, a tím i sekreci protonů do moči. Dále zvyšuje aktivitu renální glutaminázy, a tím i amoniogenezi v ledvinách. Následkem zvýšené činnosti uvedených mechanismů při hypokalémii je mírná metabolická alkalóza.
HORMONY. Hypokalémie snižuje uvolňování inzulínu a zvyšuje inzulinorezistenci, což u diabetiků 2. typu může zhoršovat hyperglykémii, a tím i kompenzaci cukrovky. Proto je potřeba aktivně pátrat po deficitu iontů draslíku u pacientů s DM i v situacích, kdy zrovna nemají diabetickou ketoacidózu. Hypokalémie rovněž snižuje sekreci aldosteronu (za předpokladu, že právě hyperaldosteronismus není onou příčinou vedoucí k hypokalémii) .
LEDVINY. Hypokalémie vede k mírné polyurii ze dvou důvodů: (1) Zvyšuje pocit žízně zvýšením hladiny angiotenzinu II v CNS; (2) Nepřímo snižuje účinnost ADH, protože narušuje aktivaci renální adenylátcyklázy, jejíž produkt cAMP je 2. poslem v antidiuretickém účinku vazopresinu. Déletrvající hypokalémie může vést k rozvoji tzv. kaliopenické nefropatie spadající do kategorie tubulointersticiálních nefritid. Funkčně se projevuje poklesem koncentrační schopnosti ledvin, polyurií a malou proteinurií. Dále je popsána vazba mezi hypokalémií při hyperaldosteronismu a vznikem renálních cyst histologicky vycházejících z epitelu sběracích kanálků. Mechanismus jejich vzniku je nejasný, korekce hypokalémie ale vede k regresi cyst.
Klasifikace hyperkalémie
Mírná hyperkalémie = plazmatická koncentrace iontů draslíku mezi 5,3 – 6,0 mmol/l.
Střední hyperkalémie = plazmatická koncentrace iontů draslíku mezi 6,0 – 7,0 mmol/l.
Těžká hyperkaleéie = plazmatická koncentrace iontů draslíku nad 7,0 mmol/l.
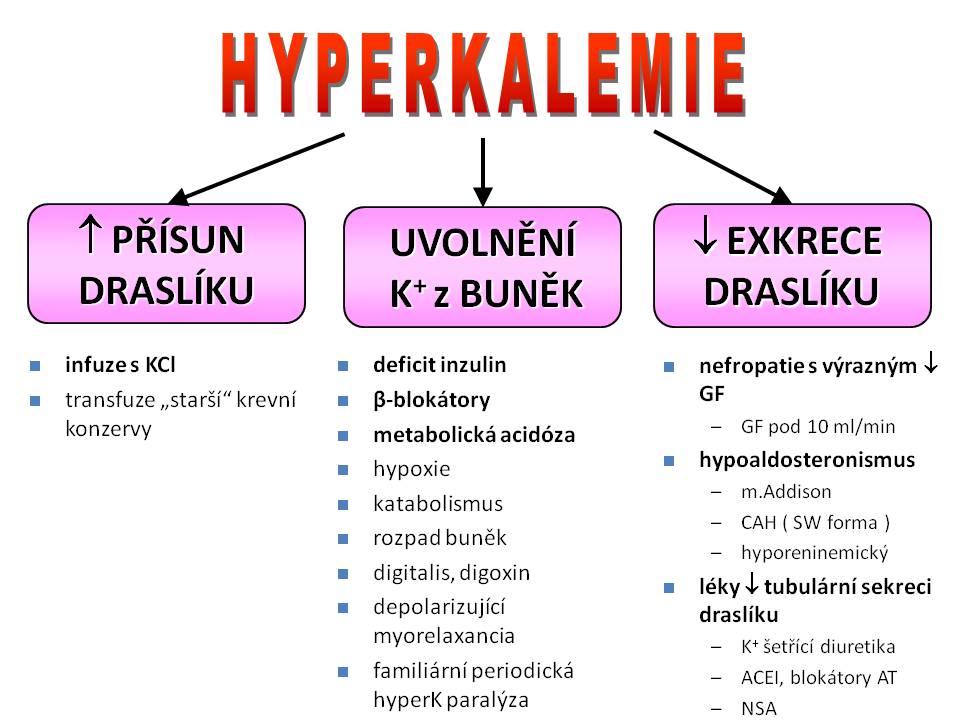
Příčiny hyperkalemie
Pseudohyperkalémie
Před tím, než přistoupíme k diferenciálně diagnostické rozvaze nad možnými příčinami hyperkalémie, je vždy potřeba u pacienta vyloučit, zda nejde o falešně pozitivní nález, tzv. pseudohyperkalémii - tedy situaci, kdy je zvýšená koncentrace iontů draslíku ve vyšetřovaném vzorku krve (nebo plazmy), ale nikoliv v krvi/plazmě v těle.
Příčiny pseudohyperkalémie uvádí následující přehled:
- Chybný odběr krve z končetiny, do které kape infúze s draselnou solí.
- Chybný odběr vzorku krve na stanovení kalémie do zkumavky určené pro odběr krevního obrazu – tyto odběrovky obsahují draselnou sůl (kalium EDTA nebo kalium citrát), které vyvázáním iontů vápníku brání krevnímu srážení, kdežto odběrovky na biochemické vyšetření základních složek plazmy typu iontů nic takového neobsahují.
- Příliš dlouhé zaškrcení nebo přiliš intenzivní „cvičení“ s končetinou, ze které je odebírán vzorek krve, což vede k úniku iontů draslíku ze svalových buněk. Obdobně vadí „ždímání“ končetiny při nevalném toku krve z jehly. Hladina kalia pak může být zkreslena až o plus 2 mmol/l.
- Vzorek je razantně „protřepán“. Tímto traumatem se uvolňují ionty draslíku z červených krvinek.
- Vzorek není centrifugován do 1 h po odběru, a tak hemolýzou dochází k uvolnění iontů draslíku jednak z erytrocytů, jednak z agregovaných trombocytů.
- Hereditární sferocytóza, popř. jiné vrozené hemolytické anémie, u kterých je snížena odolnost erytrocytů. To klade vyšší nároky na rychlost a péči při zpracování vzorku.
- Výrazná leukocytóza (nad 50 000/µl), například při leukémii, nebo výrazná trombocytóza (nad 1 000 000/µl), např. po splenektomii, kdy se draslík uvolňuje z těchto krevních elementů. Při trombocytóze se na každých 100 000 trombocytů navíc zvyšuje kalémie o 0,07 – 0,15 mmol/l.
- Chyba laboratoře.
Etiologie hyperkalémie
- Zvýšený přísun iontů draslíku do organismu;
- Snížené vylučování iontů draslíku z organismu;
- Poruchu distribuce iontů draslíku v organismu.
Zvýšený přísun iontů draslíku do organismu
Nadbytek iontů draslíku v dietě je jako „čistá“ příčina hyperkalémie prakticky vyloučen, protože, jak již bylo řečeno dříve, zdravé ledviny představují mocný nástroj regulace K+ homeostázy, díky kterému je organismus schopen se adaptovat na stav zvýšeného přísunu zvýšením exkreční frakce draselných iontů. FEK může stoupnout až na 150 – 200 %, což odpovídá schopnosti ledvin vyloučit denně 400 – 600 mmol K+. Jiná situace je u pacientů s výrazným omezením renálních funkcí. Zde výše příjmu draselných iontů přímo určuje hladinu kalémie, jejíž udržení v přijatelných mezích je potom závislé na dialýze (téma Náhrada funkce ledvin a téma Přehled umělých léčebných náhrad ledvinových funkcí).
Nepřiměřeně vysoký přísun iontů draslíku infúzí. K prevenci těchto situací se nedoporučuje překračovat koncentraci iontů draslíku 40 – 60 mmol/l v infuzním roztoku a rychlost podání 20 mmol K+ za hodinu. Pokud léčebná situace vyžaduje vyšší přívod iontů draslíku v infúzi (např. při léčbě diabetické ketoacidózy), je nutné vést infúzi s draslíkem nikoliv přes „obyčejnou“ infuzní pumpu, ale přes lineární dávkovač. Také je potřeba počítat s tím, že aplikace vyšší koncentrace iontů draslíku v roztoku je bolestivá, a takový roztok by pak měl kapat ne do periferní, ale do centrální žíly (téma Intenzivní medicína v terapii akutního renálního selhání).
Transfúze většího množství krve nebo „starší“ krevní konzervy. Krevní konzervy jsou proti krevnímu srážení ošetřeny kalium citrátem a s tím je potřeba při masívnějších transfuzích počítat. Navíc s přibývajícím „stářím“ krevní konzervy dochází k určitému stupni hemolýzy (životnost dárcovských červených krvinek v krevní konzervě je vždy kratší než obvyklých 120 dní).
Snížené vylučování iontů draslíku z organismu
Nefropatie s výrazným poklesem glomerulární filtrace. Vysoce výkonná tubulární sekrece iontů draslíku v kortikální části sběracích kanálků a v přilehlé části distálních tubulů je schopna kompenzovat i značný pokles glomerulární filtrace kalia, a tak dochází k retenci iontů draslíku až při poklesu GF pod 1/5 normálních hodnot. Hyperkalémie se obvykle objevuje až při poklesu GF pod 1/10 normy, tedy pod 10 – 15 ml/min v rámci akutního nebo chronického renálního selhání (téma Patofyziologie a klinické aspekty akutního poškození a selhání ledvin a téma Chronická ledvinová nedostatečnost a terminální stádia onemocnění ledvin).
Hlavním mechanismem renální adaptace v podobě zvýšené tubulární sekrece při klesající GF, které brání rozvoji hyperkalémie, je stoupající sekrece aldosteronu a zvyšující se aktivita Na+/K+-ATPázy v distálním nefronu. Podmínkou účinnosti aldosteronu je dostatečný přítok iontů sodíku do distálního nefronu, protože, jak už víme z předchozího výkladu, zpětná resorpce iontů sodíku vytváří příznivý elektrický gradient pro přesun draselných iontů z tubulární buňky do negativně nabitého lumina. Chemický gradient je pak udržován činností bazolaterálně umístěné Na+/K+-ATPázy, jejíž aktivita stoupá jak přímo – vlivem hyperkalémie, tak nepřímo – vlivem aldosteronu.
Další cestou přizpůsobení cest metabolismu K+ při klesající GF je intestinální adaptace se zvýšením exkrece iontů draslíku v tenkém a tlustém střevě. Tenké střevo přispívá k exkreci iontů draslíku pasivně, tím, že se v něm sníží resorpce. Aktivní exkrece iontů draslíku tlustým střevem začíná stoupat při poklesu GF na 1/3 normálních hodnot a s dalším poklesem GF se zvyšuje. Exkrece iontů draslíku enterocyty tračníku je stimulována aldosteronem. Jeho mechanismus účinku je stejný jako v tubulárních buňkách. Intestinální cesta exkrece tak ve finále může přispět k vylučování iontů draslíku z organismu dalšími asi 20 – 30 mmol/den, což při snížených renálních možnostech představuje 30 až 70 % celkové sekrece draselných iontů z těla.
Hypoaldosteronismus. Chybějící sekrece anebo snížený účinek aldosteronu může mít řadu příčin.
- Primární hypoaldosteronismus vzniká při postižení kůry nadledvin, jako je tomu u Addisonovy choroby. V tomto případě jde o autoimunitní postižení kůry nadledvin, které vede k chronickému hypokorticismu se sníženou sekrecí jak aldosteronu, tak kortizolu a nadledvinových androgenů. Akutní adrenokortikální insuficience může vzniknout při dekompenzaci u pacienta s Addisonovou nemocí, který se dostane do stresové situace (nemoc, operace atp.), anebo při akutním krvácení do nadledvin při krvácivých chorobách, při traumatu (novorozenci), nebo ve formě Waterhouse-Friderichsenova syndromu při meningokokové sepsi.
- Některé z forem kongenitální adrenální hyperplázie (CAH) jsou provázeny solnou poruchou z nedostatku aldosteronu. Nejčastěji jde o deficit 21-hydroxylázy, popř. o její SW (salt wasting) formu, kdy je nedostatečná syntéza jak gluko-, tak mineralokortikoidů a naopak je přebytek nadledvinových androgenů, které u děvčátek vedou k virilizaci genitálu a u chlapců k předčasné pubertě.
- Další příčinou může být hyporeninemický hypoaldosteronismus, kdy je snížena sekrece reninu, a tím pádem i snížená formace angiotenzinu II stimulujícího sekreci aldosteronu. Takto snížená aktivita systému RAA provází některé intersticiální nefritidy, obstrukční nefropatie a především diabetickou nefropatii. Pokud je u těchto chorob hyperkalémie provázena acidózou a není výrazněji snížena GF, mluvíme o tzv. renální tubulární acidóze IV. typu.
Léky snižující tubulární sekreci iontů draslíku. Takových farmak je celá řada. ACE inhibitory blokují angiotenzin-konvertující enzym, a tím i tvorbu angiotenzinu II a uvolňování aldosteronu. Obdobně blokátory angiotenzinových receptorů snižují syntézu aldosteronu. Cyklosporin snižuje sekreci reninu. Nesteroidní antiflogistika zase snižují sekreci reninu cestou inhibice tvorby prostaglandinů. Kalium-šetřící diuretika buď kompetují s aldosteronem v jeho vazbě na receptory v tubulárních buňkách (spironolakton), nebo ruší účinek aldosteronu blokádou kanálů ENaC v distálním nefronu s následným snížením reabsorpce iontů sodíku, a tím i exkrece iontů draslíku (amilorid, triamteren); obdobně působí i antibiotikum trimetoprim. Analogem spironolaktonu je gestagen drospirenon používaný v kombinované hormonální antikoncepci. Tento jeho antimineralokortikoidní, a tím i mírně antihypertenzivní účinek je výhodným vedlejším efektem. Heparin může u pacientů s poklesem renálních funkcí způsobit hyperkalémii svým mírně inhibičním účinkem na syntézu aldosteronu.
Poruchy distribuce iontů draslíku
Deficit inzulínu. Chybějící účinek inzulínu, který za fyziologických okolností navozuje v periferních tkáních anabolismus a podporuje vstup iontů draslíku (spolu s ionty hořčíku a s fosforečnany) do buněk, přispívá k mírné postprandiální hyperkalémii u diabetiků, ale také k významnému intracelulárnímu deficitu kalia u diabetiků v ketoacidóze.
Hypoxie. Nedostatek kyslíku ve tkáních snižuje produkci ATP, a tím snižuje i aktivitu membránové Na+/K+-ATPázy na buněčných membránách. To vede k vzestupu extracelulární koncentrace iontů draslíku.
Metabolická acidóza podporuje výměnu H+ iontů vstupujících do buněk za kationty K+ vystupující z buněk. Tento proces je výraznější u acidóz způsobených anorganickými než organickými kyselinami – vysvětlení bylo podáno v odstavcích o vztahu iontů draslíku a ABR.
Rozpad buněk. Při traumatech provázených zhmožděním svalů, při rozsáhlých hematomech, popáleninách, masivní hemolýze, ale také při rozpadu nádorových buněk vlivem onkologické léčby a při katabolických stavech (např. při stresovém katabolismu) se může uvolnit velké množství iontů draslíku z buněk. Katabolismus spojený s degradací makroergních fosfátů snižuje množství nedifuzibilních intracelulárních aniontů, které by jinak vázaly ionty draslíku uvnitř buněk.
Familiární hyperkalemická periodická paralýza je vzácná autosomálně dědičná choroba charakterizovaná náhlými atakami generalizované svalové paralýzy na podkladě distribuční hyperkalémie. Příčinou je mutace sodíkového kanálu kosterního svalstva. K záchvatům svalové slabosti obvykle dochází během odpočinku po předchozím cvičení resp. po zvýšené fyzické námaze. Epizody paralýzy trvají cca 15 – 60 minut.
Betablokátory adrenergních receptorů. Jde o léky používané při léčbě vysokého krevního tlaku, u chronického srdečního selhání anebo jako antiarytmika. Svým sympatolytickým účinkem snižují aktivitu Na+/K+-ATPázy, a tím podporují ztráty iontů draslíku z buněk ven.
Intoxikace deriváty digitalisu. Digoxin je kardiotonikum, které zvyšuje intracelulární koncentraci iontů vápníku v kardiomyocytech, a tím i sílu kontrakce. Mechanismus účinku spočívá v blokádě Na+/K+-ATPázy , což paralelně spolu s hyperkalémií vede k vzestupu koncentrace iontů sodíku uvnitř kardiomyocytů. Intracelulární přebytek iontů sodíku pak je snižován Na+/Ca2+ výměníkem. Tím dochází k vzestupu hladiny vápenatých iontů uvnitř buněk myokardu s výsledným pozitivně inotropním účinkem.
Depolarizující myorelaxancia. Patří do skupiny periferních myorelaxancií využívaných v anesteziologii ke svalové relaxaci během celkové anestézie. Příkladem je sukcinylcholin. Mechanismus působení spočívá v účinku na nikotinové receptory na postsynaptické membráně nervosvalové ploténky s následným zvýšením permeability membrány pro ionty sodíku a draslíku. V prvé fázi dochází k depolarizaci, která je ale protrahovaná a vede ke ztrátě elektrické dráždivosti svalů, ve 2. fázi dochází k částečné depolarizaci, která odpovídá za to, že jsou prvotní svalové záškuby vystřídány myorelaxací. Vzhledem k mechanismu svého účinku vedou tyto léky k výstupu draselných iontů ze svalových buněk.
Infúze argininu anebo lyzinu rovněž může vést k hyperkalémii, protože tyto aminokyseliny s kladným nábojem vstupují do buněk výměnou za draslík
Příznaky hyperkalémie
Souhrn patogeneze proarytmogenního účinku hypokalemie a hyperkalemie
- SRDCE je při hyperkalémii nejohroženějším orgánem. Hyperkalémie působí proarytmogenně. Podklad tohoto účinku stejně jako při hypokalémii spočívá ve změnách membránového potenciálu jak pracovního, tak převodního myokardu. Z elektrofyziologického hlediska hyperkalémie zkracuje třetí fázi AP a snižuje negativitu KMP, následkem čehož se zkracuje komorová repolarizace a mění excitabilita myokardu a rychlost vedení vzruchu po srdci. Nyní se na tyto důsledky podíváme blíže.
Zkrácená komorová repolarizace. Hyperkalémie zvyšuje vodivost membrány pro ionty draslíku tím, že brání inaktivaci jednoho z proudů draselných iontů zajišťujících repolarizaci. Výsledkem je rychlejší průběh komorové repolarizace, který se na EKG projeví (a) jako zkrácení a „zašpičatění“ vlny T (diferenciálně diagnosticky koronární T při infarktu myokardu jsou sice také vyšší, ale zároveň širší) a (b) celkově zkrácením intervalu QT; při těžší hyperkalémii pak depresí úseků ST.
Změny excitability a rychlosti vedení myokardu. Hyperkalémie vede ke zmenšení negativity KMP. Následkem depolarizace se nejprve při mírné hyperkalémii excitabilita zvyšuje. K vyvolání AP je za takových podmínek zapotřebí menšího vzruchu, protože se zmenšuje voltážní rozdíl mezi KMP (fyziologicky asi – 90 mV) a prahovým potenciálem pro otevření rychlých natriových kanálů (- 70 mV). Zvýšená excitabilita pak zrychluje vedení, a to se na EKG může projevit jako zkrácení intervalu PQ. Ovšem při těžší hyperkalémii se excitabilita snižuje, protože pokud se hodnota KMP depolarizací dostane nad prahový potenciál AP (tedy negativita KMP – na rozdíl od normální situace – bude větší – tedy blíže nule – než hodnota prahového potenciálu AP), snižuje se počet aktivovaných natriových kanálů, které jsou zodpovědné za rychlou depolarizaci. Rychlost depolarizace se snižuje a prodlužuje se fáze 0 akčního potenciálu. Snížená excitabilita pak zpomaluje rychlost vedení a to se na EKG při střední hyperkalémii projeví prodloužením intervalu PQ, rozšířením komplexu QRS. Oproti blokům Tawarových ramének jsou symetricky rozšířeny všechny části komplexu QRS. Při těžké hyperkalémii mizí vlna P, nicméně stimulace komor ze sinoatriálního uzlu může zůstat zachována; nastává další rozšíření QRS a jeho splynutí s vlnou T a křivka na EKG nabývá charakteru sinusoidy s frekvencí odpovídající SA uzlu - mluvíme o tzv. sinoventrikulárním rytmu. Také se mohou objevit AV bloky všech stupňů. Při velmi těžké hyperkalémii (nad 9 mmol/l) se objevuje junkční nebo idioventrikulární rytmus a bradykardie a následuje asystolie, nebo nejprve komorová fibrilace a pak asystolie.
Hodnocení hyperkalémie na EKG má svá úskalí:
- Změny na EKG nekorespondují přímo úměrně s výší hyperkalémie.
- Některé známky hyperkalémie jsou nespecifické, což může vést k jejich přehlédnutí.
- Přechod z normálního nálezu na EKG do komorové arytmie či asystolie může být velmi rychlý!!
KOSTERNÍ SVALY. Mírná hyperkalémie mechanismem částečné depolarizace zvyšuje nervosvalovou dráždivost, což se může projevit brněním rtů anebo prstů a svalovými záškuby. Při těžší hyperkalémii se ale nervosvalová dráždivost snižuje, a to vede k ascendentně postupující svalové slabostí, ztrátě svalového napětí až paralýze. Postižení respiračních svalů překvapivě nebývá natolik zásadní, aby ohrozilo život pacienta respirační insuficiencí.
HLADKÉ SVALSTVO. Stejně jako u svalů kosterních vede i v případě hladkého svalstva zažívacího traktu mírná hyperkalémie ke zvýšení nervosvalové dráždivosti, což se může projevit jako abdominální křeče a průjem. Při těžší hyperkalémii se pak motilita GIT snižuje.
ACIDOBAZICKÁ ROVNOVÁHA. Hyperkalémie v proximálním tubulu snižuje reabsorpci bikarbonátů, v kortikální části sběracích kanálků inhibuje sekreci protonů do moči a také snižuje tvorbu amoniaku. Následkem aktivace uvedených mechanismů je mírná metabolická acidóza.
HORMONY. Hyperkalémie zvyšuje sekreci aldosteronu (za předpokladu, že příčinou hyperkalémie není hypoaldosteronismus).
Léčba hyperkalémie
- jsou přítomny EKG známky anebo
- sérová hladina kalia v přesahuje 6,5 mmol/l.
Elektrická stabilizace myokardu. Toho lze dosáhnout intravenózním podáním iontů vápníku obvykle ve formě calcium gluconicum. Výše kalcémie totiž ovlivňuje hodnotu prahového potenciálu, při kterém se otevírají napěťově řízené natriové kanály odpovídající za rychlou fázi depolarizace. Léčbou navozená hyperkalcémie posunuje prahový potenciál do méně negativních hodnot, což by za normokalemické situace snižovalo excitabilitu, ale při těžké hyperkalémii to obnovuje normální excitabilitu – alespoň částečně se obnoví voltážní rozdíl mezi klidovým membránovým potenciálem membrány depolarizované hyperkalémií a prahem pro spuštění AP. Tím se zvýší „síla“ depolarizačního proudu iontů natria a rychlost fáze 0 akčního potenciálu. Účinek podaných iontů kalcia se dostavuje během 2 – 3 minut a přetrvává asi 30 až 60 minut.
Přesun iontů draslíku do buněk. Pro tento krok je lékem první volby inzulín, který stimuluje Na+/K+-ATPázu na membránách buněk a tím zajistí přesun K+ iontů do intracelulárního kompartmentu. Inzulín se podává intravenózně a jako prevence hypoglykémie je současně v infúzi podávána glukóza. Účinek lze očekávat během 20 – 30 minut a přetrvává u „rychlých“ inzulínů asi 4 – 6 hodin. Výjimkou jsou pacienti s hyperglykémií, kde stačí samotný inzulín. Jako doplňkovou léčbu k inzulínu lze přidat β2-sympatomimetika (např. bronchodilatans salbutamol), která rovněž aktivují Na+/K+-ATPázu. Jejich cesta podání závisí na dostupné formě léku, tedy buď intravenózně, nebo lze i inhalačně, ale je nutná mnohem vyšší dávka.
Vyloučení přebytku iontů draslíku z organismu. Jde o léčebný krok vhodný jen pro hyperkalémie spojenou s retencí iontů draslíku. U pacientů s vysoce sníženou GF volíme hemodialýzu. U ostatních perorálně podáváme iontoměniče – pryskyřice, jako např. polystyrensulfonát sodný, z kterého se v zažívacím traktu uvolňují kationty sodíku a vážou kationty draslíku.
Použitá lietaruta a literatura k dalšímu studiu
- ASSADI F. Diagnosis of hypokalemia: a problem-solving approach to clinical cases. Iran J Kidney Dis. 2008 Jul;2(3):115-22
- BERNE RB, LEVY MN, KOEPPEN BM, STANTON BA. Physiology, 5th edition, Mosby 2004, pp. 22 – 30, 685 – 693
- GENNARI FJ. Hypokalemia. N Engl J Med. 1998 Aug 13;339(7):451-8
- GENNARI FJ, Segal AS. Hyperkalemia: An adaptive response in chronic renal insufficiency. Kidney Int. 2002 Jul;62(1):1-9
- GIEBISH G, HEBERT SC, WANG WH. New aspects of renal potassium transport. Pflugers Arch. – Eur J Physiol. 2003 Jun;446(3):289-97
- GUYTON AC, HALL JE. Textbook of medical physiology, 11th edition, Elsevier Saunders 2006, pp. 365 – 368
- HOLLANDER-RODRIGUEZ JC, CALVERT JF Jr. Hyperkalemia. Am Fam Physician. 2006 Jan 15;73(2):283-90
- JABOR A a kol. Vnitřní prostředí. 1.vydání, Grada 2008, str. 45 – 55
- JANG HY, McDONOUGH AA. Recent advancest in understanding integrative kontrol of potassium homeostasis. Annu. Rev. Physiol. 2009, 71: 381 – 401
- KUSUMOTO F, ECG Interpetation: From patophysiology to clinical application, Springer Science + Business Media, LLC 2009, pp. 249 – 253
- LEHNHARDT A, KEMPER MJ. Pathogenesis, diagnosis and management of hyperkalemia. Pediatr Nephrol. 2011 Mar;26(3):377-84
- LIM S. Approach to hypokalemia. Acta Med Indones. 2007 Jan-Mar;39(1):56-64.
- McCANCE KL, HUETHER SE, BRASHERS VL, ROTE NS. Pathophysiology: the biological basis for disease in adults and children, 6th edition, Mosby Elsevier 2010, pp. 106- 111
- MUSSO CG. Potassium metabolism in patiens with chronic kidney dinase. Int Urol Nephrol. 2004; 36(3): 465 – 472
- NORRIS KC, LEVINE B, GANESAN K. Thyreotoxic periodic paralysis associated with hypokalemia and hypophosphatemia. Am J Kidney Dis. 1996 Aug;28(2):270-3
- OSADCHII OE. Mechanisms of hypokalemia-induced ventricular arrhythmogenicity. Fundam Clin Pharmacol. 2010 Oct;24(5):547-59
- PAJEREK J, ŠTOLCOVÁ A, FIALKA J. Otrava kaliem. Pediatr. Pro Praxi 2006; 1: 49-51
- PARHAM WA, Mehdirad AA, Biermann KM, Fredman CS. Hyperkalemia revisited. Tex Heart Inst J. 2006;33(1):40-7
- RYŠAVÁ R. Hypokalémie. Interní Med. 2006; 9: 385 – 388
- RYŠAVÁ R. Hypokalémie u renálních tubulopatií. Med. Pro Praxi 2008; 5(4): 146 – 149
- SCHÜCK O. Poruchy metabolismu vody a elektrolytů v klinické praxi. Grada, Praha 2000, str. 107-131
- SNYDERS DJ. Structure and fiction of cardiac potassium channels. Cardiovascular Res. 1999 May;42(2):377-90
- UNWIN RJ, LUFT FC, SHIRLEY DG. Pathophysiology and management of hypokalemia: a clinical perspective. Nat Rev Nephrol. 2011 Feb;7(2):75-84.
- VESELÝ J. Tlaková diuréza a arteriální hypertenze. Epava 2002, str. 34 – 37, 61 – 73, 133 – 134, 248 – 250
- WEINER ID, WINGO CS. Hypokalemia-consequences, causes, and correction. J Am Soc Nephrol. 1997 Jul;8(7):1179-88
- WEINER ID, WINGO CS. Hyperkalemia: a potential silent killer. J Am Soc Nephrol. 1998 Aug;9(8):1535-43

